温馨提示:请使用火狐或者Chrome的网页浏览器来查看报告
微科盟细菌完成图(基于ONT测序)云流程结题报告
一 概述
细菌完成图(Complete Genome)代表细菌基因组组装的最高标准,其核心要求为闭合环状序列的完整性——即无 Gap、无 N 碱基,并完整保留染色体与质粒的全部遗传信息。然而,二代测序技术受限于短读长,在面对细菌基因组内高度重复的 rRNA 操纵子簇(16S-23S-5S)、插入序列(IS 元件)以及高 GC 含量区域时,常因读长无法覆盖重复单元全长而导致组装中断,形成大量 Contig 碎片。传统补救方案依赖 PCR 扩增结合 Sanger 测序逐一定点填补 Gap,流程繁琐且难以保证全覆盖。相较之下,Nanopore 超长读长技术能够一次性贯穿上述复杂区域,从根本上规避了短读长组装中的重复序列错配和结构重建偏差,从而在无需填补操作的前提下,一次性获得真实、连续且完整的该菌株基因组骨架结构。
二 项目流程
2.1 测序实验流程
2.1.1 基因组DNA提取与检测
采用SDS(十二烷基硫酸钠)法或STE(蔗糖-Tris-EDTA)缓冲液法提取样本基因组DNA,随后利用琼脂糖凝胶电泳检测DNA的纯度与完整性,并使用Qubit荧光计对DNA浓度进行精确定量。
2.1.2 Nanopore文库构建与测序
取经电泳检测合格的DNA样品,使用Covaris超声波破碎仪将其随机打断至适宜片段长度。处理完成后的DNA片段,采用Nanopore测序平台配套建库试剂盒,经末端修复、加测序接头、纯化等步骤完成文库制备。文库构建完成后,先使用Qubit荧光计进行初步定量,并将文库稀释至适宜浓度;随后使用Agilent 2100生物分析仪(Bioanalyzer)检测文库插入片段(insert size)大小,确认符合预期后,采用实时荧光定量PCR(q-PCR)法对文库的有效浓度进行准确定量,以确保文库质量合格。最终将构建好的文库于Nanopore测序平台完成上机测序。

2.2 生信分析流程

2.2.1 原始数据质量控制
测序下机后,使用NanoFilt对原始下机数据(raw reads)进行质量控制,过滤低质量及短片段序列,以获得高质量的有效数据(clean reads),确保后续组装和分析的准确性。
2.2.2 基因组组装、抛光与评估
采用Flye长读长组装软件对过滤后的有效数据进行基因组从头组装(de novo assembly),并通过Medaka基于原始测序信号进行碱基级抛光(polish),以提高基因组单碱基准确度。组装完成后,使用DnaApler将组装序列的起始位点重定向至复制起始蛋白基因(dnaA),确保基因组序列以规范的起点为开端。利用QUAST(Quality Assessment Tool for Genome Assemblies)对组装结果进行全面的质量评估(包括基因组大小、GC含量、N50、基因数量等指标),通过CheckM对组装基因组进行物种鉴定及完整性评估,并使用SAMtools depth分析基因组各碱基位点的测序覆盖深度,以评估组装区域的覆盖均一性。
2.2.3 基因组结构及功能注释
在基因组完成质量评估后,使用Prokka(Prokaryotic Genome Annotation)对基因组编码序列(CDS)、tRNA及rRNA等结构进行快速自动化注释。随后,利用eggNOG-mapper进行直系同源蛋白聚类及功能注释,同时使用ABRicate对基因组携带的毒力因子、耐药基因及分泌系统等特定功能组分进行鉴定。
2.2.4 基因组可视化
整合上述组装与注释信息,使用Circlize(R包)绘制环形基因组图(Circos plot),以可视化展示基因组的结构特征、功能分布及覆盖度等信息。
三 数据分析结果
牛津纳米孔测序技术(Oxford Nanopore Technology,ONT)测序技术的核心组件主要包括纳米孔(nanopore)和马达蛋白(motor protein)(Wang et al., 2021)。纳米孔嵌入于电阻膜中,作为核酸分子通过的微小通道,其作用在于允许单链DNA或RNA分子依次穿过,并实时检测核酸序列穿过时引起的电流变化,通过不同碱基(A、T、C、G)所产生的特征性电流信号差异实现对序列的碱基识别。马达蛋白则起到驱动和调控作用,其结合于核酸链末端,引导核酸链以可控的速度逐位通过纳米孔,确保电流信号的稳定采集和碱基识别的准确性。二者协同工作,共同实现了ONT平台对长片段核酸分子的高通量、实时测序。
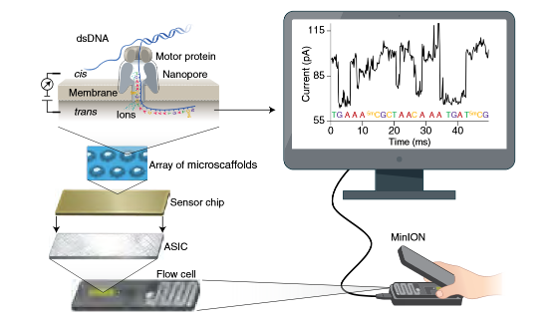
3.1 原始数据质量控制
01.qc/
├── 01.nanoplot_before_filter
│ └── *
│ ├── LengthvsQualityScatterPlot_kde.html
│ ├── NanoStats.txt
│ └── Non_weightedHistogramReadlength.html
├── 02.porechop
│ └── *
│ └── *.filtered.fastq.gz
├── 03.filtlong
│ └── *
│ └── *.filtered.fastq.gz
└── 04.nanoplot_after_filter
└── *
├── LengthvsQualityScatterPlot_kde.html
├── NanoStats.txt
└── Non_weightedHistogramReadlength.html
测序下机后,使用Porechop(Bonenfant et al., 2022)对原始下机数据(raw reads)进行接头识别与切除,并利用Filtlong(Petersen et al., 2022)对reads进行质量过滤,去除低质量及短片段序列,以获得高质量的有效数据(clean reads),确保后续组装和分析的准确性。为评估质控效果,分别于质控前后使用NanoPlot对reads的长度分布、碱基质量分布等特征进行可视化统计。
3.1.1 数据质控可视化
核密度估计图能够直观展示reads在“长度-平均质量”二维平面上的分布密度,颜色越深表示该区域reads数量越多。
图1 质控前reads长度与平均质量的核密度估计图
横坐标为read长度(bp,对数坐标),纵坐标为平均碱基质量(Q-score)。图中可见质控前存在大量短片段和低质量reads,主密度区域位于长度500–5000 bp、质量7–15的范围内。
图2 质控后reads长度与平均质量的核密度估计图
横坐标与纵坐标同图1。与图1对比,质控后短于500 bp及平均质量低于Q7的reads被有效去除,高密度区域向更长长度(>5 kb)和更高质量(>10)集中,整体分布更加紧凑。
长度分布直方图显示了reads按不同长度区间的频数分布,用于评估质控前后reads长度的变化趋势。
图3 质控前reads长度分布图
横坐标为read长度区间(bp),纵坐标为reads数量(或频率)。质控前reads长度呈现明显的右偏分布,大量reads集中在0–2 kb区间,长片段(>10 kb)占比较低。
图4 质控后reads长度分布图
横坐标与纵坐标同图3。质控后短片段比例显著下降,长度分布整体向右平移,主峰向1–3 kb或更长区域移动,长片段占比相对增加。
3.1.2 质控统计汇总
表1汇总了质控前后reads的各项核心统计量,用于定量评估质控效果。
| before | after | |
|---|---|---|
| Mean read length | 10355.9 | 12494.2 |
| Mean read quality | 15.6 | 17.2 |
| Median read length | 9245.0 | 10433.0 |
| Median read quality | 17.7 | 18.3 |
| Number of reads | 10000.0 | 7381.0 |
| Read length N50 | 12513.0 | 12902.0 |
| STDEV read length | 8210.0 | 6759.5 |
| Total bases | 103559187.0 | 92219653.0 |
表1 质控前后reads核心统计量汇总
注:质控后reads数量减少,但长度均值、N50和碱基平均质量均预期上升,表明低质量短片段被有效剔除,数据整体质量得到提升。
表2按照不同的Q-score阈值(Q10、Q20、Q25、Q30)对质控前后reads进行分层统计,包括符合该质量阈值的reads数量占比及对应的碱基总数。
| before_num | after_num | before_percent | after_percent | before_base | after_base | |
|---|---|---|---|---|---|---|
| Q10 | 9595.0 | 7376.0 | 96.0% | 99.9% | 99.5Mb | 92.1Mb |
| Q15 | 7907.0 | 6488.0 | 79.1% | 87.9% | 81.5Mb | 77.8Mb |
| Q20 | 907.0 | 1117.0 | 9.1% | 15.1% | 10.3Mb | 13.2Mb |
| Q25 | 1.0 | 2.0 | 0.0% | 0.0% | 0.0Mb | 0.0Mb |
| Q30 | 0.0 | 0.0 | 0.0% | 0.0% | 0.0Mb | 0.0Mb |
表2 质控前后不同质量分层的统计
注:Q30表示平均质量≥30的reads,其碱基准确率≥99.9%。质控后各质量层级的占比和碱基总数预期显著提高,尤其在高阈值(Q25、Q30)区域改善更为明显。
3.2 基因组组装与校正
02.assemble/
├── 01.flye
│ └── *
│ ├── assembly.fasta
│ └── assembly_info.txt
├── 02.medaka
│ └── *
│ └── consensus.fasta
└── 03.dnaapler
└── *
├── dnaapler_all_reorientation_summary.tsv
└── dnaapler_reoriented.fasta
3.2.1 该步骤分析方法
3.2.1.1 基因组组装
采用Flye组装软件(Kolmogorov et al., 2019),基于重叠-布局-共识(Overlap-Layout-Consensus,OLC)算法,对质控后的有效数据进行基因组从头组装(de novo assembly)。Flye是针对单分子长读长测序数据(包括Nanopore)专门设计的组装工具,通过构建重叠图并识别高置信度的contig路径,能够在不打断重复序列的前提下生成连续、接近完整的组装结果,充分发挥了Nanopore超长读长在跨越基因组重复区域的优势。
3.2.1.2 组装结果抛光
Nanopore测序数据中插入/缺失(InDel)错误的比例显著高于碱基替换错误,约占总错误的3%–7%。这类错误会破坏基因的开放阅读框(ORF),导致下游基因预测和功能注释出现大量移码突变、假基因或错误编码序列,严重影响注释质量。因此,组装完成后使用Medaka对初始组装结果进行碱基级抛光(polish)。Medaka是一款基于循环神经网络(RNN)的抛光工具,利用原始Nanopore读长对应的电流信号事件特征,能够高效、准确地识别并修正插入/缺失错误,显著提高组装序列的碱基一致性准确度。
3.2.1.3 基因组起始位点重定向
细菌基因组通常为环状DNA分子,在序列数据库中习惯将复制起始区域(通常以dnaA基因作为标志)的特定碱基位置定义为序列起点(坐标1),以便于不同菌株基因组之间的比较、共线性分析和注释。然而,组装软件输出的线性化contig其起始点是任意的。因此,使用DnaApler(Bouras et al., 2024)对抛光后的基因组序列进行重定向处理,通过识别contig上的dnaA基因(或其同源类似基因),自动将基因组序列进行旋转和重定向,使dnaA基因的第一个碱基成为整个序列的起点,从而满足细菌基因组标准化展示和比较的需求。
3.2.2 组装结果统计
经Flye组装、Medaka抛光及DnaApler定向后的最终基因组统计如下表所示:
| Contig | Gene_Reoriented | Top_Hit | Coverage | Identity_Percentage | |
|---|---|---|---|---|---|
| 0 | contig_1 | dnaA | sp~A7Z0C3~DNAA_BACVZ | 100.0 | 99.78 |
| 1 | contig_2 | No_MMseqs2_hits | No_MMseqs2_hits | No_MMseqs2_hits | No_MMseqs2_hits |
表3 基因组组装与定向结果统计
注:Contig:序列名称;Gene_Reoriented:是否成功将dnaA基因置于序列起点;Top_Hit:起始基因的具体名称;Coverage:定量评估找到的基因覆盖参考序列的比例,越接近1代表基因越完整,可信度越高;Identity_Percentage:定量评估匹配到的基因与参考序列的氨基酸同源性,越高说明匹配越可靠。
3.3 组装结果评估
03.assemble_qc/
├── 01.quast
│ └── *
│ ├── report.html
│ └── report.tsv
├── 02.checkM
│ └── *
│ └── storage
│ └── bin_stats_ext.tsv
└── 03.depth
└── *
└── *.coverage.txt
3.3.1 组装质量评估(QUAST)
QUAST(Quality Assessment Tool for Genome Assemblies)(Gurevich et al.,
2013)是一种通用的基因组组装质量评估工具,可计算组装序列的多项质量指标,涵盖连续性、准确性和完整性等方面。与CheckM通过标记基因评估基因组进化完整性不同,QUAST侧重于从序列组装本身的角度评估组装的物理质量,两者互为补充。
QUAST的运行分为两种模式:无参考基因组模式下,仅计算不需要参考序列的指标,包括contig数量、总长度、N50/L50、GC含量等基础统计指标,以及通过reads回比评估覆盖度的相关信息,但无法提供准确性相关的评估(如misassembly检测)。基于参考基因组模式下,在提供高质量近缘参考基因组时,QUAST可进行更全面的评估,将组装序列与参考基因组进行比对,计算基因组覆盖度(Genome
fraction)、重复率(Duplication ratio)、比对长度(Largest alignment / Total aligned
length)、NA50/NGA50等高级指标,并检测misassembly(错误组装)。本次评估采用无参考基因组模式(因目标菌株无合适的近缘参考基因组),重点评估组装序列的连续性与基本特征,准确性方面的评估由后续的samtools
depth和基因注释分析补充。
QUAST评估结果如下:
| dnaapler_reoriented | |
|---|---|
| Assembly | |
| # contigs (>= 0 bp) | 2 |
| # contigs (>= 1000 bp) | 2 |
| # contigs (>= 5000 bp) | 2 |
| # contigs (>= 10000 bp) | 1 |
| # contigs (>= 25000 bp) | 1 |
| # contigs (>= 50000 bp) | 1 |
| Total length (>= 0 bp) | 4242507 |
| Total length (>= 1000 bp) | 4242507 |
| Total length (>= 5000 bp) | 4242507 |
| Total length (>= 10000 bp) | 4237487 |
| Total length (>= 25000 bp) | 4237487 |
| Total length (>= 50000 bp) | 4237487 |
| # contigs | 2 |
| Largest contig | 4237487 |
| Total length | 4242507 |
| GC (%) | 45.85 |
| N50 | 4237487 |
| N75 | 4237487 |
| L50 | 1 |
| L75 | 1 |
| # N's per 100 kbp | 0.00 |
表4 QUAST组装质量评估结果
注:各指标含义如下:
contigs(≥ X bp):长度大于或等于特定阈值(0 bp、1000 bp、5000 bp、10000 bp、25000 bp、50000
bp)的contig数量,该值随阈值升高而减少,可反映组装片段的长短分布。
Total length(≥ X bp):长度大于或等于特定阈值的contig的碱基总数,与完整基因组大小越接近,说明组装覆盖越全面。
contigs:最终组装获得的所有contig总数(无长度筛选),数量越少(理想情况为1),表明组装连续性越好。
Largest contig:所有contig中长度最大者的碱基数,对于环状细菌基因组,若成功环化且无质粒,该值应等于总长度。
Total length:所有contig的碱基总和,即组装的基因组预估总大小。
GC(%):全基因组中鸟嘌呤(G)和胞嘧啶(C)碱基所占的百分比,GC含量是物种特征性指标,显著偏离预期值可能提示污染或组装错误。
N50:将所有contig从长到短排序后,累加长度首次达到总长度50%时对应contig的长度,N50越高代表组装连续性越好。
N75:与N50类似,累加长度达到总长度75%时的contig长度,反映了较短contig端的组装质量。
L50:达到N50所需的最小contig数量,L50越小(理想值为1),说明少数长contig即可覆盖基因组一半的长度。
L75:达到N75所需的最小contig数量,同样反映组装的长片段覆盖效率。
N's per 100 kbp:每100千碱基(100,000 bp)序列中不确定碱基“N”的平均个数,该值越接近0,说明组装中缺口(gap)越少,序列完整性越高。
3.3.2 组装质量评估(CheckM)
细菌基因组组装完成后,获得的contigs序列可能存在多种质量问题,包括:污染(混入了其他微生物如共生菌、噬菌体或实验室污染的序列)、不完整(缺失部分基因或基因组区域)、碎片化(组装不连续,被错误打断为多个小片段)以及碱基错误(GC含量异常、序列内存在大量模糊碱基N)。这些问题会直接影响后续的基因注释、比较基因组学、代谢通路重建等分析的可靠性。传统方法(如仅依据N50、覆盖度等)无法准确评估基因组的完整性和污染程度。CheckM(Parks et al., 2015)通过分析基因组中普遍存在的单拷贝标记基因(marker genes),能够定量评估基因组的完整性、污染率,并提供丰富的统计指标(GC含量、N50、编码密度等),帮助用户筛选出高质量(High Quality, HQ)和中等质量(Medium Quality, MQ)的bins,是当前微生物基因组质量评估的金标准工具之一。
| bin_id | dnaapler_reoriented |
|---|---|
| marker lineage | g__Bacillus |
| Completeness | 99.481328 |
| Contamination | 0.0 |
表5 CheckM基因组质量评估结果
注:CheckM对每个基因组bin输出以下主要质量指标:
marker lineage:基因组的标记谱系;
Completeness:完整性(%),即保守单拷贝标记基因被检出的比例,越高越好,高质量通常要求≥90%;
Contamination:污染率(%),即标记基因出现多拷贝的比例,越低越好,高质量要求≤5%。
3.3.3 基于samtools depth的基因组覆盖度分析
在基因组组装完成后,利用samtools depth(Danecek et al., 2021)统计组装基因组每个位点的测序覆盖深度,是评估组装质量与测序数据利用效率的关键步骤,其主要用途包括:评估组装完整性(覆盖深度均匀、无大面积零深度区域,表明组装序列与原始测序数据匹配良好,没有明显的缺失或错误重复);检测潜在组装错误(深度剧烈波动如局部极低或极高可能提示重复序列区域深度异常高、污染序列或嵌合体深度异常低或缺失、单拷贝区域与多拷贝区域区分不清);指导后续矫正与过滤(通过深度分布可确定合理的深度阈值,用于过滤低覆盖区域、识别高拷贝重复单元、辅助评估杂合区域);量化测序数据冗余(平均覆盖深度反映了测序数据量相对于基因组大小的富余程度,为后续变异检测、基因注释等提供基础质控指标)。

图5 各contig沿基因组位置的覆盖深度分布
注:横轴表示基因组上的位点坐标(bp),纵轴表示该位点的测序覆盖深度。不同颜色的折线代表不同的contig(或染色体)。覆盖深度的波动可直接反映组装序列与测序读段的匹配均匀性:平坦的折线提示覆盖均匀,组装质量较好;剧烈尖峰或深谷则可能对应重复序列(深度过高)、组装错误或缺失区域(深度过低)。若某contig出现大段深度为零的区域,表明该段序列可能为组装错误引入的虚假序列,或未被测序数据覆盖的缺口。当多个contig共存于同一坐标轴时,可通过比较纵轴数值判断各序列的相对拷贝数或污染情况(例如质粒contig通常具有与染色体相近或数倍的覆盖深度)。
表6 各contig覆盖深度统计汇总
注:基于samtools depth输出的每个位点覆盖深度,按contig分组计算以下统计量:
min:该contig上所有位点的最低覆盖深度,若为0说明存在完全无读段覆盖的位点;
max:最高覆盖深度,用于识别潜在的重复序列区域或异常富集区;
mean:平均覆盖深度,反映该contig整体的测序冗余水平;
median:中位数覆盖深度,比均值更稳健,当均值远高于中位数时,提示存在少量极高覆盖位点(如重复单元)拉高了平均值;
std(标准差):衡量覆盖度的离散程度,标准差越大说明覆盖度在contig内部波动越剧烈,均匀性越差。
比较不同contig的均值:若某contig的均值显著低于总体均值,可能为污染序列、外源DNA或组装错误引入的冗余片段;若显著高于总体均值,则可能对应高拷贝质粒或重复区域。结合min与max可进一步判断是否存在极端异常位点。
3.4 基因组的结构注释
04.annotation/01.prokka/
└── *
├── *_prokka.faa
├── *_prokka.ffn
├── *_prokka.gff
├── *_prokka.tsv
└── *_prokka.txt
基因组结构注释(Genome
Annotation)是基因组学研究的核心环节之一,其根本目的是将原始的基因组DNA序列转化为具有生物学意义的解读。在获得基因组测序与组装结果之后,得到的仅为由A、T、C、G构成的碱基序列,这些序列本身并不直接揭示其生物学功能。基因组注释的任务正是在这些序列上识别出所有具有生物学意义的特征,包括编码蛋白质的基因(CDS)、非编码RNA基因(如rRNA、tRNA、tmRNA)、启动子区域、重复序列等,并为它们标注相应的功能信息。具体而言,进行基因组结构注释具有以下几方面的重要意义:
揭示基因组的编码潜能:通过注释可明确基因组中蛋白质编码基因的数量及其在基因组上的具体位置、长度、方向等信息,从而了解该生物体的基本基因组成;
为功能研究提供基础:结构注释是功能注释的前提,只有先确定了基因的位置和序列,才能进一步通过序列比对、结构域分析等手段推测其生物学功能,注释结果是后续比较基因组学、泛基因组分析、代谢通路重建、进化研究等一切深入分析的基础数据;
满足数据提交与共享的标准化需求:国际公共数据库(如GenBank、ENA、DDBJ)要求提交的基因组数据必须包含标准的注释信息,规范的注释能够确保数据的可重复利用和跨研究之间的可比较性;
提升研究效率与可扩展性:传统的人工注释或基于网页的在线注释服务耗时较长(数小时至数天),难以满足大规模测序数据的高通量分析需求,自动化注释工具能够在保证注释质量的前提下大幅提升效率,使研究者能够快速获得基因组概览,并方便地整合到自动化分析流程中。
3.4.1 Prokka原理简介
Prokka是由澳大利亚墨尔本大学的Torsten
Seemann开发的一款专用于原核生物(细菌、古菌及病毒)基因组的快速注释工具。它本质上是一个“包装器”(wrapper),整合了多种已有的优秀软件工具,避免了重复开发。Prokka的注释流程主要分为以下几个步骤:
输入与准备:Prokka接受预组装的基因组DNA序列(FASTA格式)作为输入,可以是完成图(无gap的完整序列),也可以是de
novo组装产生的scaffold序列。用户可通过命令行参数指定输出目录、文件前缀、物种分类(细菌/古菌/病毒等)以及菌株的属、种、株名称等信息。
蛋白质编码基因(CDS)的预测:Prokka使用Prodigal软件来识别基因组上的开放阅读框(ORF)。Prodigal采用动态规划方法,通过预测核糖体结合位点、分析GC含量和六核苷酸分布特征,并经过多次迭代优化,来精确定位基因的起始密码子和终止密码子。Prodigal对ORF的识别设定了90
bp的长度下限。这一步骤是Prokka注释流程的核心,决定了后续所有功能注释的质量。
非编码RNA基因的识别:除了蛋白质编码基因,Prokka还会识别基因组中的非编码RNA基因,包括tRNA(转运RNA)、rRNA(核糖体RNA)以及tmRNA(转移-信使RNA)。这些RNA分子在蛋白质合成和基因表达调控中发挥着不可或缺的作用。
功能注释:在识别出基因的编码区域之后,Prokka通过将预测的蛋白质序列与多个蛋白质或蛋白质结构域数据库进行相似性比对,来预测每个基因编码产物的功能。这一过程为每个基因赋予功能描述(product
description),为后续的功能分类和代谢通路分析提供依据。
输出生成:Prokka会在约10分钟内完成一个典型细菌基因组的注释,并生成多种标准格式的输出文件,主要包括:.gff(GFF3格式的完整注释文件,可在Artemis或IGV等基因组浏览器中查看)、.gbk(GenBank格式的注释文件)、.fna(原始输入序列)、.faa(预测蛋白序列)、.ffn(预测基因核苷酸序列)、.txt(注释统计摘要)以及.log(运行日志)。这些文件既可直接用于下游分析,也可经过少量调整后提交至国际公共数据库。
3.4.2 结果展示
本次使用Prokka对目标基因组进行了全面的结构注释,获得了完整的基因集及其功能标注。以下从基因长度分布和基因类型构成两个方面对注释结果进行展示与分析。
3.4.2.1 基因长度分布

图6 基因长度分布柱状图
注:横轴为基因长度(bp),纵轴为对应长度区间的基因数量。Prokka预测的所有基因(含CDS及各类RNA基因)的长度分布呈现以下特征:峰值区间方面,基因长度分布呈现明显的单峰特征,绝大多数基因的长度集中在200–400
bp区间,这一分布模式与原核生物基因长度的典型特征一致——原核生物的蛋白质编码基因通常较短,平均长度在300 bp左右(约100个氨基酸);长尾分布方面,分布图右侧呈现较长的拖尾,存在少量长度超过1000
bp甚至2000 bp的基因,这些长基因通常编码具有多结构域的大分子蛋白质,如某些酶类、转运蛋白或调控蛋白;短基因的存在方面,分布图左侧可以看到少量长度在100–200
bp的基因,Prodigal对ORF的识别设定了90
bp的下限,因此极短基因不会被纳入预测结果,短基因中可能包含一些小型调控蛋白或信号肽编码序列,但需注意过短的预测基因也可能存在一定的假阳性风险;基因组编码密度的反映方面,基因长度的整体分布反映了该基因组的编码密度特征,若大多数基因集中在较短的区间,说明基因组较为紧凑、基因间区较少,这是原核生物基因组的普遍特征。
3.4.2.2 基因类型构成

图7 Prokka注释结果中不同类型基因的数量与占比饼图
注:包括蛋白质编码基因(CDS)、核糖体RNA(rRNA)、转运RNA(tRNA)和转运-信使RNA(tmRNA)四类。
3.5 基因组的功能注释
04.annotation/02.eggnog/
└── *
├── *.bigg_anno.tsv
├── *.cazy_anno.tsv
├── *.cog_anno.tsv
├── *.emapper.annotations
├── *.go_anno.tsv
├── *.kegg_anno.tsv
├── *.pfam_anno.tsv
└── *.total_anno.tsv
3.5.1 功能注释概述
在完成基因组结构注释、获得所有编码基因(CDS)的核酸及氨基酸序列之后,需要进一步对每个基因编码产物进行功能注释,以揭示其生物学功能(Huerta-Cepas et al.,
2017)。功能注释的核心原理是将预测得到的蛋白质序列与公共数据库中已知功能的蛋白质或蛋白质结构域进行序列相似性比对,通过同源性推断来赋予未知基因以功能描述。本研究采用eggNOG-mapper(Cantalapiedra
et al., 2021)对Prokka预测的蛋白序列进行功能注释。eggNOG-mapper是基于eggNOG(evolutionary genealogy of genes: Non-supervised
Orthologous
Groups,直系同源蛋白聚类数据库)开发的自动化功能注释工具,通过将查询序列与预计算的直系同源群进行比对,快速、准确地推断基因的功能信息。相较于传统的BLAST+GO注释方法,eggNOG-mapper利用预先构建的直系同源聚类和系统发育信息,能够提供更准确的跨物种功能转移注释,显著降低假阳性率。
eggNOG-mapper注释结果包含丰富的信息字段,主要包括:query(查询序列ID,即Prokka预测的基因ID)、seed_ortholog(预测到的直系同源群标识符)、evalue(比对期望值,用于评估比对结果的显著性)、score(比对得分,表示序列间的相似程度)、eggNOG_OGs(直系同源群编号)、max_annot_lvl(与直系同源群关联的分类学信息)、COG(Clusters
of Orthologous Groups编号,用于描述基因和蛋白质的进化关系)、description(序列的功能描述概述)、Preferred_name(基因符号,gene symbol)、GOs(Gene
Ontology注释,涵盖分子功能、细胞组分和生物过程三个维度)、EC(Enzyme Commission编号,用于标识酶的催化功能)、KEGG_ko(KEGG
Orthology标识符)、KEGG_Pathway(代谢通路注释)、KEGG_Module(代谢模块注释)、KEGG_Reaction(参与的生物反应注释)、KEGG_rclass(反应类别注释)、BRITE(BRITE层次分类注释)、KEGG_TC(Transporter
Classification编号,膜转运蛋白分类)、CAZy(Carbohydrate-Active
enZYmes,碳水化合物活性酶库注释,包括能催化碳水化合物降解、修饰及生物合成的相关酶系家族)、BiGG_Reaction(BiGG数据库的生化反应注释)以及PFAMs(蛋白质家族数据库注释,将具有相似结构和功能的蛋白质归类到相同家族)。
3.5.2 注释结果统计
对上述注释结果中常用的数据库类别——包括COG(直系同源蛋白簇)、GO(基因本体论)、EC(酶委员会编号)、KEGG_ko(KEGG直系同源标识符)、CAZy(碳水化合物活性酶)、BiGG_Reaction(BiGG生化反应)及PFAMs(蛋白质家族)——分别统计其注释到的基因数量,并采用UpSet图进行可视化展示,以直观呈现各数据库注释基因的重叠与分布情况。

图8 功能注释结果UpSet图
注:横轴展示了各数据库的注释基因数量及不同数据库间注释基因的交集情况,纵轴表示对应交集或并集的大小。UpSet图相较于传统的Venn图在处理多集合(超过4-5组)时具有更好的可读性,能够清晰展示各数据库注释的覆盖范围及其重叠关系,从而全面评估基因组功能注释的覆盖广度和各数据库之间的一致性。
3.6 基因结构分析
04.annotation/03.abricate/
└── *
├── *.bacmet2.tsv
├── *.card.tsv
├── *.ncbi.tsv
├── *.plasmidfinder.tsv
├── *.vfdb.tsv
└── *.victors.tsv
3.6.1 ABRicate原理简介
ABRicate是一款用于批量筛查基因组序列(contigs)中抗菌药物耐药(Antimicrobial Resistance, AMR)基因或毒力基因的工具,由Torsten
Seemann开发。其核心原理是将组装后的基因组序列与内置的多个功能基因数据库进行BLAST比对,从而快速鉴定基因组中携带的耐药基因、毒力因子及质粒相关基因等特定功能组分。
ABRicate的主要特点包括:仅支持组装后的contigs序列(FASTA、GenBank或EMBL格式),不支持原始的FASTQ测序读段;仅检测获得性耐药基因,无法识别由点突变引起的耐药性;采用DNA序列数据库而非蛋白质数据库进行比对。其输出结果详细,包括基因名称、在基因组上的起始/终止坐标、方向、覆盖度(%COVERAGE)、碱基一致性(%IDENTITY)、匹配数据库来源、基因产物描述及耐药表型等信息。
ABRicate内置了多个常用的功能基因数据库,可根据研究需求灵活选用:
- 耐药基因数据库:NCBI AMRFinderPlus、CARD、Resfinder、ARG-ANNOT、MEGARES
- 毒力基因数据库:VFDB(Virulence Factor Database)、Ecoli_VF
- 质粒相关数据库:PlasmidFinder
- 血清型抗原数据库:EcOH(大肠杆菌O/H抗原分析)
3.6.2 结果展示
本研究采用ABRicate对组装完成的基因组序列进行扫描,分别选用VFDB数据库鉴定毒力基因、CARD/Resfinder数据库鉴定耐药基因,以及PlasmidFinder数据库鉴定质粒相关复制子,以全面评估该菌株基因组中携带的功能组分。
3.7 基因组环形可视化
为直观展示基因组的结构特征与注释信息的空间分布,本研究采用CGview(Grant and Stothard, 2008)绘制环形基因组图(Circos plot)。Circos图以环状形式将基因组的多维信息整合于同一视图中,从外圈至内圈依次展示以下内容:
图9 基因组环形可视化图(Circos plot)
注:由外至内各圈层分别表示基因组坐标、CDS分布、非编码RNA位点、COG功能分类、覆盖深度及GC含量/GC偏移。GC偏移正值(G>C,正链)偏向复制起始方向,负值(C>G,负链)偏向复制终止方向,可用于辅助定位复制起始点(通常位于GC偏移由负转正处)。
四 参考文献
- Bonenfant, Q., Noé, L., Touzet, H., 2022. Porechop_ABI: discovering unknown adapters in Oxford Nanopore Technology sequencing reads for downstream trimming. Bioinforma. Adv. 3, vbac085. https://doi.org/10.1093/bioadv/vbac085
- Bouras, G., Grigson, S.R., Papudeshi, B., Mallawaarachchi, V., Roach, M.J., 2024. Dnaapler: A tool to reorient circular microbial genomes. J. Open Source Softw. 9, 5968. https://doi.org/10.21105/joss.05968
- Cantalapiedra, C.P., Hernández-Plaza, A., Letunic, I., Bork, P., Huerta-Cepas, J., 2021. eggNOG-mapper v2: Functional Annotation, Orthology Assignments, and Domain Prediction at the Metagenomic Scale. Mol. Biol. Evol. 38, 5825–5829. https://doi.org/10.1093/molbev/msab293
- Danecek, P., Bonfield, J.K., Liddle, J., Marshall, J., Ohan, V., Pollard, M.O., Whitwham, A., Keane, T., McCarthy, S.A., Davies, R.M., Li, H., 2021. Twelve years of SAMtools and BCFtools. GigaScience 10, giab008. https://doi.org/10.1093/gigascience/giab008
- Grant, J.R., Stothard, P., 2008. The CGView Server: a comparative genomics tool for circular genomes. Nucleic Acids Res. 36, W181–W184. https://doi.org/10.1093/nar/gkn179
- Gurevich, A., Saveliev, V., Vyahhi, N., Tesler, G., 2013. QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075. https://doi.org/10.1093/bioinformatics/btt086
- Huerta-Cepas J., Forslund K., Coelho L.P., Szklarczyk D., Jensen L.J., von Mering C., Bork P., 2017. Fast Genome-Wide Functional Annotation through Orthology Assignment by eggNOG-Mapper. Mol. Biol. Evol. 34, 2115–2122. https://doi.org/10.1093/molbev/msx148
- Kolmogorov, M., Yuan, J., Lin, Y., Pevzner, P.A., 2019. Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 37, 540–546. https://doi.org/10.1038/s41587-019-0072-8
- Parks, D.H., Imelfort, M., Skennerton, C.T., Hugenholtz, P., Tyson, G.W., 2015. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055. https://doi.org/10.1101/gr.186072.114
- Petersen, C., Sørensen, T., Westphal, K.R., Fechete, L.I., Sondergaard, T.E., Sørensen, J.L., Nielsen, K.L., 2022. High molecular weight DNA extraction methods lead to high quality filamentous ascomycete fungal genome assemblies using Oxford Nanopore sequencing. Microb. Genomics 8, 000816. https://doi.org/10.1099/mgen.0.000816
- Wang, Yunhao, Zhao, Y., Bollas, A., Wang, Yuru, Au, K.F., 2021. Nanopore sequencing technology, bioinformatics and applications. Nat. Biotechnol. 39, 1348–1365. https://doi.org/10.1038/s41587-021-01108-x