微科盟宏转录组数据分析结题报告
一 概述
地球生物圈中,微生物扮演着极为重要的角色,它们的活动影响着自然环境的营养循环, 土壤肥力,有机质的分解,以及物种与能量之间的交换。人类对微生物的研究从 Antonivan Leeuwenhoek 发明显微镜开始的数百年中,主要基于纯培养的研究方式, 而在数以万亿计的微生物种类中,仅 0.1%~1%的物种可培养,极大地限制了对微生物多样性资源的研究和开发。 宏转录组学(Metatranscriptomics)兴起于宏基因组之后,从整体水平上研究某一特定环境, 特定时期群体生命全部基因组转录情况以及转录调控规律,它以生态环境中的全部RNA 为研究对象, 避开了微生物分离培养困难的问题,能有效的扩展微生物资源的利用空间。2006 年, Leiniger 等首次使用 454 测序技术对一个复杂微生物群落的宏转录组进行研究。与宏基因组学相比较, 宏转录组学能从转录水平研究复杂微生物群落变化,能更好的挖掘潜在的新基因。
近年来,随着测序技术和信息技术的快速发展,利用新一代测序技术(Next Generation Sequencing)研究宏转录组, 能快速准确的得到大量生物数据和丰富的微生物研究信息,从而成为研究微生物多样性和群落特征的重要手段。 如致力于研究微生物与人类疾病健康关系的人体微生物组计划(HMP, Human Microbiome Project, http://www.hmpdacc.org/ ), 研究全球微生物组成和分布的全球微生物组计划(EMP, Earth Microbiome Project,http://www.earthmicrobiome.org/ ) 都主要利用高通量测序技术进行研究。
二 项目流程
2.1 测序实验流程
从RNA样品提取到最终数据获得,样品检测、建库、 测序等每一环节都会直接影响数据的数量和质量, 从而影响后续信息分析的结果。为从源头保证测序数据准确可靠, 我们承诺在数据的所有生产环节都严格把关, 从根源上确保高质量数据的产出。建库测序的流程图如下(图2.1):
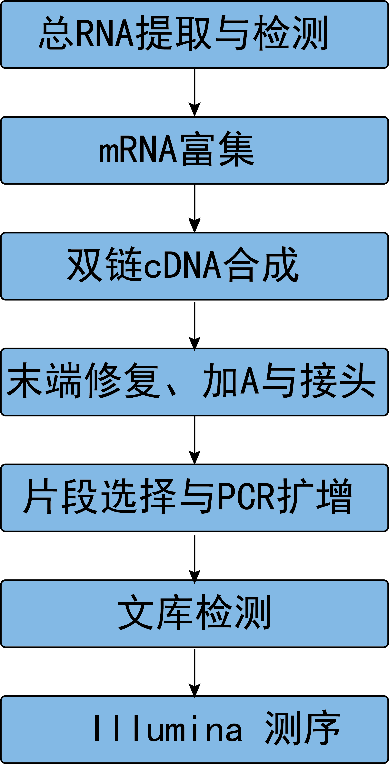
图2.1 RNA-Seq建库测序流程
2.1.1 RNA提取与检测
采用标准提取方法从组织或细胞中提取RNA,随后对RNA样品进行严格质控,质控标准主要包括以下4个方面:
- 琼脂糖凝胶电泳:分析样品RNA完整性及是否存在DNA污染;
- NanoPhotometer spectrophotometer:检测RNA纯度(OD260/280及OD260/230比值);
- Qubit2.0 Fluorometer:RNA浓度精确定量;
- Agilent 2100 bioanalyzer:精确检测RNA完整性。
2.1.2 文库构建与质检
文库构建
mRNA的获取主要有两种方式:一是利用真核生物大部分mRNA都带有polyA尾的结构特征, 通过Oligo(dT)磁珠富集带有polyA尾的mRNA。二是从总RNA中去除核糖体RNA, 从而得到mRNA。随后在NEB Fragmentation Buffer中用二价阳离子将得到的mRNA随机打断, 按照NEB普通建库方式或链特异性建库方式进行建库。(图2.2)。
- NEB普通建库:以片段化的mRNA为模版,随机寡核苷酸为引物, 在M-MuLV逆转录酶体系中合成cDNA第一条链,随后用RNaseH降解RNA链, 并在DNA polymerase I 体系下,以dNTPs为原料合成cDNA第二条链。 纯化后的双链cDNA经过末端修复、加A尾并连接测序接头(1), 用AMPure XP beads筛选200bp左右的cDNA,进行PCR扩增并再次使用AMPure XP beads纯化PCR产物, 最终获得文库。建库原理如下面左图所示。
- 链特异性建库:逆转录合成cDNA第一条链方法与NEB普通建库方法相同, 不同之处在于合成第二条链时,dNTPs中的dTTP由dUTP取代,之后同样进行 cDNA末端修复、加A尾、连接测序接头和长度筛选,然后先使用USER酶 降解含U的cDNA第二链再进行PCR扩增并获得文库。链特异性文库具有诸多优势, 如相同数据量下可获取更多有效信息;能获得更精准的基因定量、定位与注释信息; 能提供反义转录本及每一isoform中单一exon的表达水平。建库原理如下面右图所示。
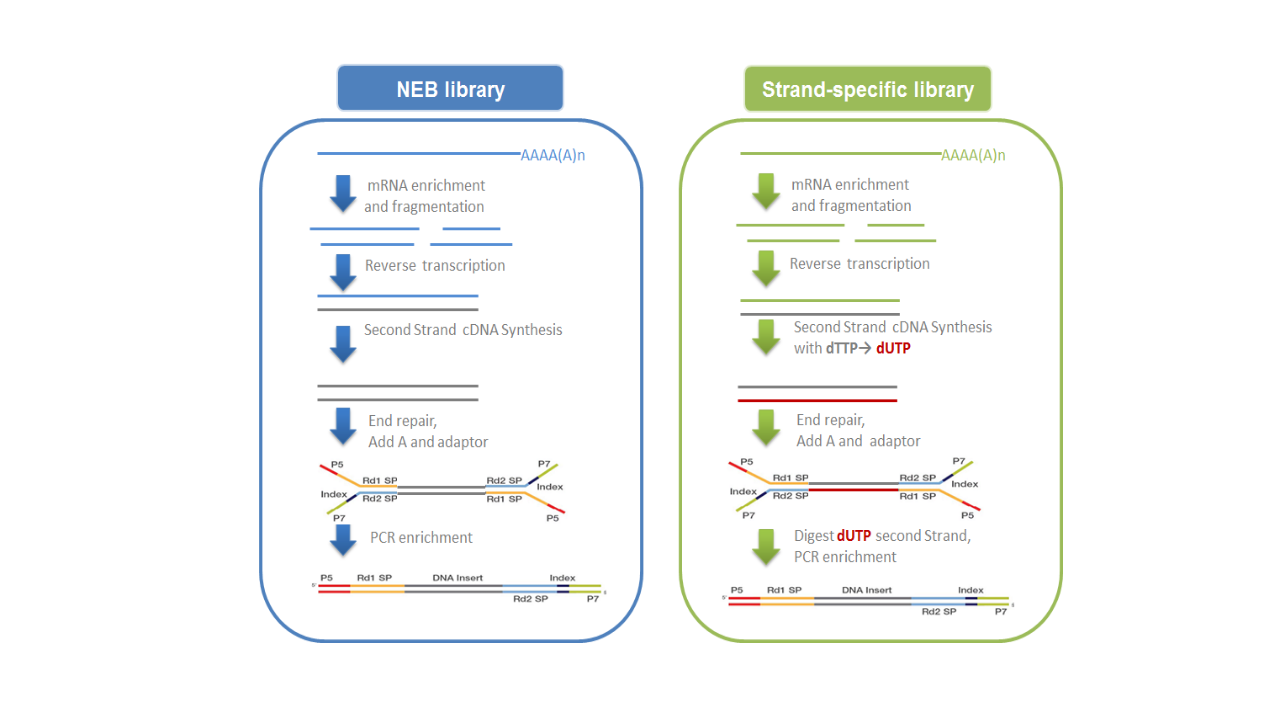
图2.2 文库构建原理示意图
注:测序接头:包括P5/P7,index和Rd1/Rd2 SP三个部分(如上图所示)。 其中P5/P7是PCR扩增引物及flow cell上引物结合的部分, index提供区分不同文库信息的Rd1/Rd2,SP即read1/read2 sequence primer, 是测序引物结合区域,测序过程理论上由Rd1/Rd2 SP向后开始进行。
文库质检
文库构建完成后,先使用Qubit2.0 Fluorometer进行初步定量, 稀释文库至1.5ng/ul,随后使用Agilent 2100 bioanalyzer 对文库的insert size进行检测,insert size符合预期后, qRT-PCR对文库有效浓度进行准确定量(文库有效浓度高于2nM),以保证文库质量。
2.1.3 上机测序
库检合格后,把不同文库按照有效浓度及目标下机数据量的需求pooling后进行Illumina测序。 测序的基本原理是边合成边测序(Sequencing by Synthesis)。 在测序的flow cell中加入四种荧光标记的dNTP、DNA聚合酶以及接头引物进行扩增, 在每一个测序簇延伸互补链时,每加入一个被荧光标记的dNTP就能释放出相对应的荧光, 测序仪通过捕获荧光信号,并通过计算机软件将光信号转化为测序峰, 从而获得待测片段的序列信息。测序过程如下图所示。
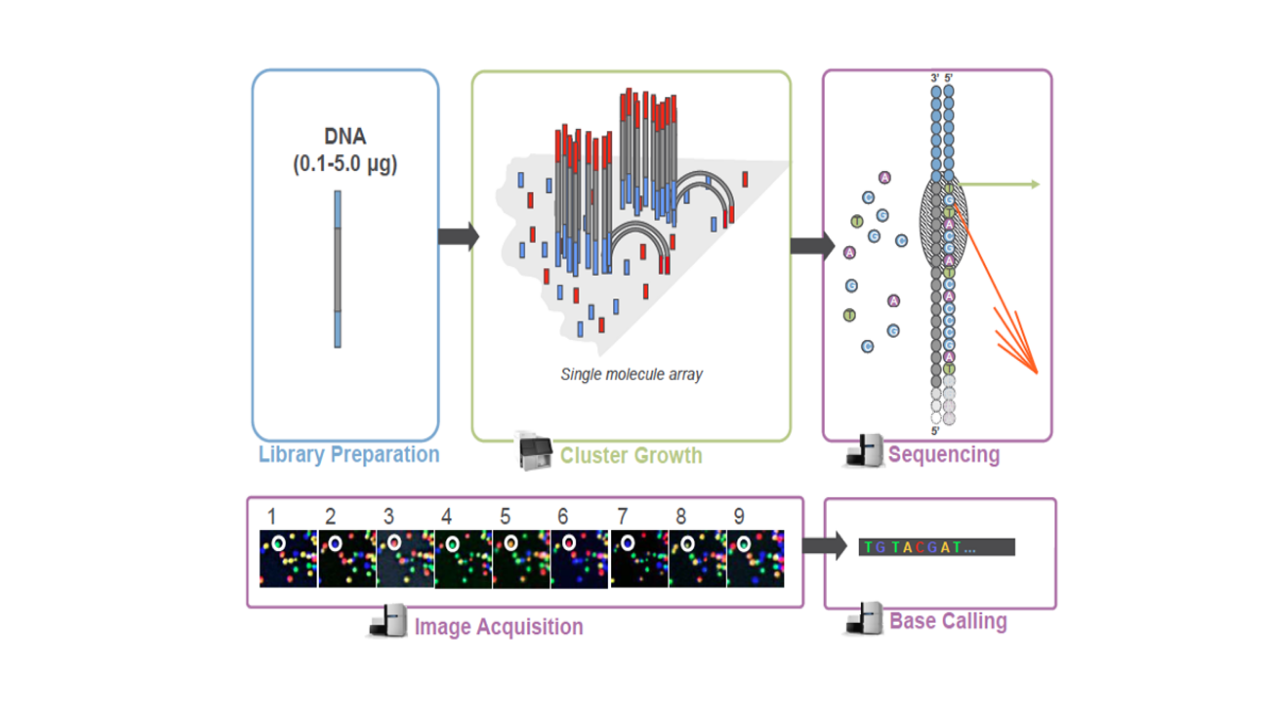
图2.3 Illumina测序原理示意图
2.2 生信分析流程
总的来说,在对原始数据进行预处理(质控去宿主,去rRNA), 转录本拼接,基因去冗余,非冗余基因定量, 获得基因丰度表之后,有三条分析路线:1. 挑选差异表达基因,差异基因富集分析,差异蛋白互作网络分析; 2. 结合非冗余基因功能注释结果(KEGG, GO, CARD, CAZy, EggNOG)进行功能定量,然后进行功能基础统计,差异比较,相关性分析; 3. 结合非冗余基因物种注释结果进行物种定量,然后进行物种基础统计,差异比较,相关性分析,多样性分析(α多样性,β多样性)
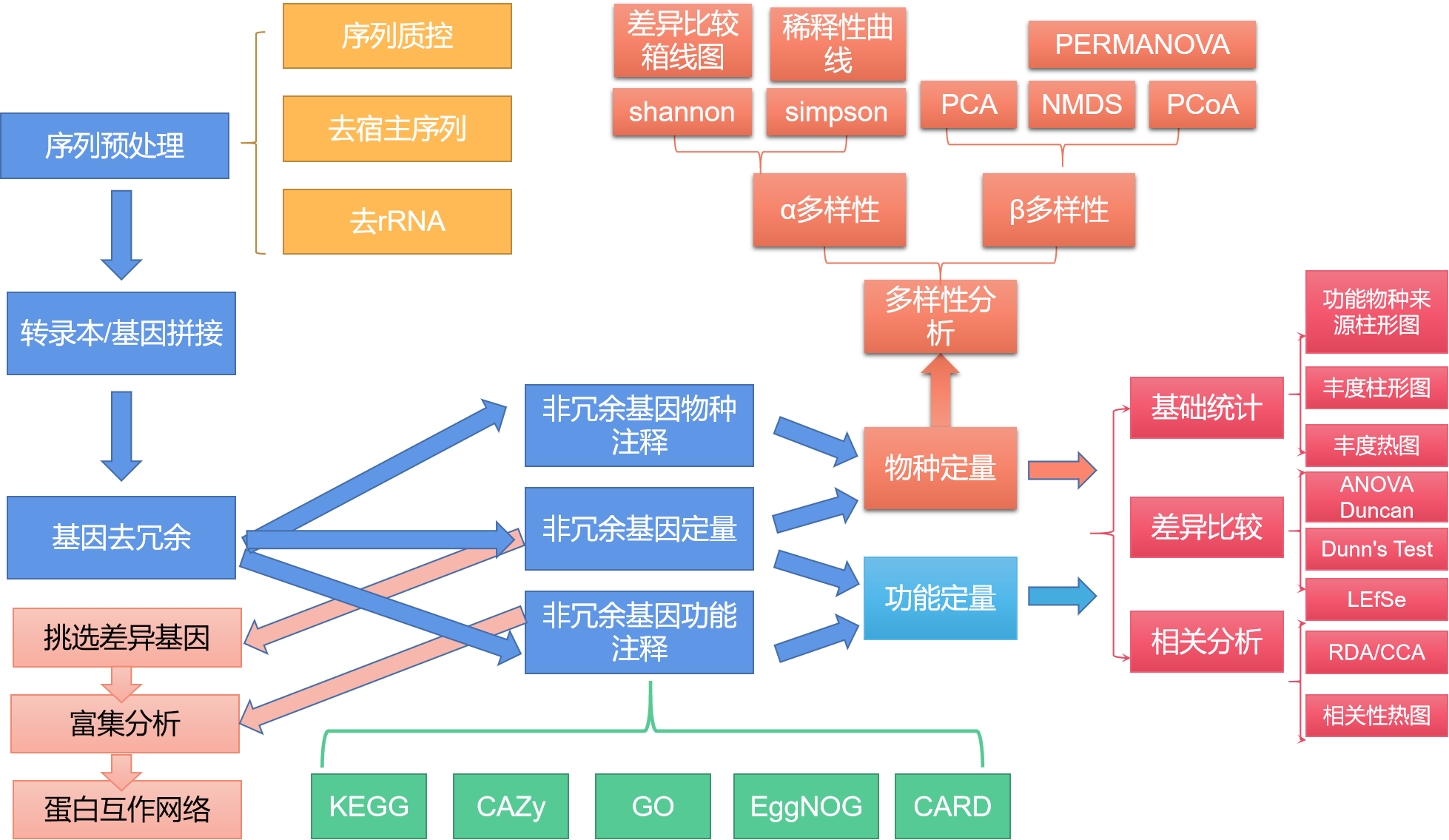
图2.4 宏转录组分析技术流程
三 结果文件夹解读
3.1 测序数据预处理
01.QC/ ├── Sample*_fastp.html [各样本fastp软件质控报告] ├── summary_before_filtering.xls [各样本质控前质量情况汇总] ├── summary_after_filtering.xls [各样本质控后质量情况汇总] ├── summary_after_remove_host.xls[各样本去宿主序列后的质量情况汇总] ├── summary_after_sortmerna.xls [各样本去rRNA序列后的质量情况汇总]
质控
用fastp软件[1]对每一个样本的测序数据raw data做质控处理(参数: --overrepresentation_analysis --trim_front1 2 --trim_front2 2 --cut_front --cut_tail --cut_window_size 3 --cut_mean_quality 30),得到clean data用于下游分析,质控规则如下:
- 对整条read做保留或弃去处理
- 根据碱基质量值弃去低质量的整条read
- 当一条read中N碱基的个数达到一定的数量(默认为5个)时,弃去整一条read;
- 当一条read中低质量碱基(默认质量值低于15的碱基为低质量碱基)达到一定的比例(默认这个比例值为40%)时,弃去整一条read。
- 根据read长度弃去低质量的整条read
- 当一条read的碱基个数少于一定的数量(默认为15个碱基)时,弃去整一条read。
- 对read做局部裁剪
- 强制性删除头部和尾部各2个碱基
- 因为测序反应一般起始和结束阶段的碱基质量是最差的,所以我们统一裁剪掉read1和read2它们各自的5'端的2个碱基、3'端的2个碱基。
- 根据碱基质量值删除滑动窗口内的碱基
- 一般来说,测出来的read,开头的碱基质量差,然后随着测序反应的稳定,碱基质量会逐步上升,后面随着反应时间的延长、化学产物的积累、酶受到的环境不利影响的积累,到后面碱基质量会下降,所以需要对read的开头和结尾的低质量碱基区做裁剪。
- 我们以3个碱基的长度为一个窗口的大小,从read的头部向尾部滑动,计算窗口内的3个碱基的平均质量值,如果平均质量值小于阈值30,那么表明当前窗口的碱基质量差,就删除这个窗口内的所有的3个碱基,然后滑动到下一组的3个碱基,计算平均质量值,做类似的删除处理;如果当前的窗口的平均质量值大于或等于30,那么表明当前窗口的碱基质量足够好了,于是停止滑动窗口,保留这个窗口以及后面的所有碱基。同理,从read的尾部向头部,也做类似的滑动窗口删除碱基的处理。
- adapter序列裁剪
- 默认情况下,对pair-end 的read1和read2做两序列比对,查看比对结果中,read1的右端是否会超出read2的右端,read2的左端是否会超出read1的左端,如果超出的话,表明是测到了adapter序列,则会把超出的部分给剪掉。
将所有样本的质控信息做提取整合,生成总体的样本测序数据质控信息统计表。
表3-1-1 各样本质控前质量情况汇总(summary_before_filtering.xls)
| sample id | total reads | total bases | total giga bases | q20 bases | q30 bases | q20 rate | q30 rate | read1 mean length | read2 mean length | gc content |
|---|---|---|---|---|---|---|---|---|---|---|
| Sample01 | 94494288 | 14174143200 | 14.17G | 13895050677 | 13398282623 | 0.980310 | 0.945262 | 150 | 150 | 0.615763 |
| Sample02 | 75920308 | 11388046200 | 11.39G | 11148321804 | 10714603532 | 0.978949 | 0.940864 | 150 | 150 | 0.599634 |
| Sample03 | 82491774 | 12373766100 | 12.37G | 12018717200 | 11422591274 | 0.971306 | 0.923130 | 150 | 150 | 0.588121 |
| Sample04 | 71941226 | 10791183900 | 10.79G | 10488926927 | 9984248239 | 0.971990 | 0.925223 | 150 | 150 | 0.593806 |
| Sample05 | 74314402 | 11147160300 | 11.15G | 10878233678 | 10393127082 | 0.975875 | 0.932356 | 150 | 150 | 0.571716 |
| Sample06 | 90955308 | 13643296200 | 13.64G | 13212380625 | 12522866969 | 0.968416 | 0.917877 | 150 | 150 | 0.599757 |
| Sample07 | 62262952 | 9339442800 | 9.34G | 9170680360 | 8844062750 | 0.981930 | 0.946958 | 150 | 150 | 0.502165 |
| Sample08 | 63985774 | 9597866100 | 9.6G | 9426226308 | 9090113070 | 0.982117 | 0.947097 | 150 | 150 | 0.473198 |
| Sample09 | 67789804 | 10168470600 | 10.17G | 9962236303 | 9569345445 | 0.979718 | 0.941080 | 150 | 150 | 0.471983 |
| Sample10 | 66649444 | 9997416600 | 10.0G | 9750119167 | 9289749136 | 0.975264 | 0.929215 | 150 | 150 | 0.511303 |
| Sample11 | 65553170 | 9832975500 | 9.83G | 9591411799 | 9128565669 | 0.975433 | 0.928362 | 150 | 150 | 0.476220 |
| Sample12 | 66995322 | 10049298300 | 10.05G | 9803695215 | 9332937555 | 0.975560 | 0.928715 | 150 | 150 | 0.474125 |
- sample id: 样本ID
- total reads: 样本序列总数
- total bases: 样本碱基总数
- total giga bases: 样本碱基总数(G为单位)
- q20 bases: 质量分高于20(错误率0.01)的碱基总数
- q30 bases: 质量分高于30(错误率0.001)的碱基总数
- q20 rate: 质量分高于20(错误率0.01)的碱基占比(Q20)
- q30 rate: 质量分高于30(错误率0.001)的碱基占比(Q30)
- read1 mean length: read1平均长度
- read2 mean length: read2平均长度
- gc content: GC含量
表3-1-2 各样本质控后质量情况汇总(summary_after_filtering.xls)
| sample id | total reads | total bases | total giga bases | q20 bases | q30 bases | q20 rate | q30 rate | read1 mean length | read2 mean length | gc content |
|---|---|---|---|---|---|---|---|---|---|---|
| Sample01 | 94068998 | 13652808352 | 13.65G | 13423411838 | 12954879011 | 0.983198 | 0.948880 | 145 | 145 | 0.614272 |
| Sample02 | 75519604 | 10950331285 | 10.95G | 10757697625 | 10350111731 | 0.982408 | 0.945187 | 145 | 144 | 0.598190 |
| Sample03 | 81968842 | 11855815809 | 11.86G | 11568085748 | 11008621313 | 0.975731 | 0.928542 | 144 | 144 | 0.586258 |
| Sample04 | 71455496 | 10337136582 | 10.34G | 10095060125 | 9622001692 | 0.976582 | 0.930819 | 144 | 144 | 0.591949 |
| Sample05 | 73976470 | 10715257938 | 10.72G | 10493874125 | 10035535526 | 0.979339 | 0.936565 | 144 | 144 | 0.569931 |
| Sample06 | 90305616 | 13028724443 | 13.03G | 12681284685 | 12039786720 | 0.973333 | 0.924096 | 144 | 144 | 0.598273 |
| Sample07 | 62086004 | 8928990341 | 8.93G | 8790796899 | 8484463575 | 0.984523 | 0.950215 | 143 | 143 | 0.499385 |
| Sample08 | 63808118 | 9195816862 | 9.2G | 9055021352 | 8738358668 | 0.984689 | 0.950254 | 144 | 144 | 0.470155 |
| Sample09 | 67614300 | 9763364590 | 9.76G | 9590880383 | 9219548386 | 0.982334 | 0.944300 | 144 | 144 | 0.469150 |
| Sample10 | 66411566 | 9533266437 | 9.53G | 9327503545 | 8897203010 | 0.978416 | 0.933280 | 143 | 143 | 0.509021 |
| Sample11 | 65389006 | 9410559617 | 9.41G | 9204012975 | 8766996679 | 0.978052 | 0.931613 | 144 | 143 | 0.473464 |
| Sample12 | 66824124 | 9632202871 | 9.63G | 9421999997 | 8976746700 | 0.978177 | 0.931952 | 144 | 143 | 0.471570 |
- sample id: 样本ID
- total reads: 样本序列总数
- total bases: 样本碱基总数
- total giga bases: 样本碱基总数(G为单位)
- q20 bases: 质量分高于20(错误率0.01)的碱基总数
- q30 bases: 质量分高于30(错误率0.001)的碱基总数
- q20 rate: 质量分高于20(错误率0.01)的碱基占比(Q20)
- q30 rate: 质量分高于30(错误率0.001)的碱基占比(Q30)
- read1 mean length: read1平均长度
- read2 mean length: read2平均长度
- gc content: GC含量
去除宿主序列
为了避免宿主序列对后续分析的影响,使用 BWA[2](参数: mem –k 30)软 件把 质控后的 reads 与宿主比对,去除比对成功长度高于 reads 总长 80%的reads。
去除宿主序列之后的质量信息统计表格如下:
表3-1-2 各样本去除宿主序列后质量情况汇总(summary_after_remove_host.xls)
| sample id | total reads | total bases | total giga bases | q20 bases | q30 bases | q20 rate | q30 rate | read1 mean length | read2 mean length | gc content |
|---|---|---|---|---|---|---|---|---|---|---|
| Sample01 | 92660400 | 13453514206 | 13.45G | 13234985292 | 12778006497 | 0.983757 | 0.949789 | 145 | 145 | 0.612408 |
| Sample02 | 74823094 | 10852390618 | 10.85G | 10665352231 | 10263967998 | 0.982765 | 0.945779 | 145 | 145 | 0.597897 |
| Sample03 | 81282798 | 11760816762 | 11.76G | 11480839811 | 10928977669 | 0.976194 | 0.929270 | 144 | 144 | 0.586163 |
| Sample04 | 70958352 | 10268854035 | 10.27G | 10032323245 | 9564752287 | 0.976966 | 0.931433 | 144 | 144 | 0.592158 |
| Sample05 | 73171606 | 10603716465 | 10.6G | 10390254255 | 9940110245 | 0.979869 | 0.937418 | 144 | 145 | 0.569393 |
| Sample06 | 88820094 | 12822403279 | 12.82G | 12491051950 | 11865492237 | 0.974158 | 0.925372 | 144 | 144 | 0.597633 |
| Sample07 | 61557742 | 8859546933 | 8.86G | 8726123984 | 8424257817 | 0.984940 | 0.950868 | 143 | 143 | 0.498843 |
| Sample08 | 63407958 | 9143096568 | 9.14G | 9005762934 | 8692533425 | 0.984980 | 0.950721 | 144 | 144 | 0.469848 |
| Sample09 | 67257714 | 9715929916 | 9.72G | 9546459752 | 9178300773 | 0.982557 | 0.944665 | 144 | 144 | 0.469085 |
| Sample10 | 65806112 | 9453660527 | 9.45G | 9253013310 | 8828076149 | 0.978776 | 0.933826 | 143 | 143 | 0.508154 |
| Sample11 | 65021182 | 9361553856 | 9.36G | 9158125916 | 8724505024 | 0.978270 | 0.931951 | 144 | 143 | 0.473201 |
| Sample12 | 66421060 | 9578315142 | 9.58G | 9371405753 | 8929825110 | 0.978398 | 0.932296 | 144 | 143 | 0.471304 |
- sample id: 样本ID
- total reads: 样本序列总数
- total bases: 样本碱基总数
- total giga bases: 样本碱基总数(G为单位)
- q20 bases: 质量分高于20(错误率0.01)的碱基总数
- q30 bases: 质量分高于30(错误率0.001)的碱基总数
- q20 rate: 质量分高于20(错误率0.01)的碱基占比(Q20)
- q30 rate: 质量分高于30(错误率0.001)的碱基占比(Q30)
- read1 mean length: read1平均长度
- read2 mean length: read2平均长度
- gc content: GC含量
去除rRNA
使用SortMeRNA[3](参数:默认参数)软件去除reads中的rRNA序列( mRNA, tRNA会被保留,它们都是有重要功能的 )。
去除rRNA序列之后的质量信息统计表格如下:
表3-1-2 各样本去rRNA序列后的质量情况汇总(summary_after_sortmerna.xls)
| sample id | total reads | total bases | total giga bases | q20 bases | q30 bases | q20 rate | q30 rate | read1 mean length | read2 mean length | gc content |
|---|---|---|---|---|---|---|---|---|---|---|
| Sample01 | 92125548 | 13376467034 | 13.38G | 13159817762 | 12705802093 | 0.983804 | 0.949862 | 145 | 145 | 0.612802 |
| Sample02 | 74298984 | 10777257717 | 10.78G | 10592637961 | 10194668557 | 0.982870 | 0.945943 | 145 | 145 | 0.598538 |
| Sample03 | 80715244 | 11679845046 | 11.68G | 11403323536 | 10856021884 | 0.976325 | 0.929466 | 144 | 144 | 0.586799 |
| Sample04 | 70484138 | 10201179117 | 10.2G | 9967425303 | 9503558748 | 0.977086 | 0.931614 | 144 | 144 | 0.592796 |
| Sample05 | 72340446 | 10484472466 | 10.48G | 10274567638 | 9830037878 | 0.979979 | 0.937581 | 144 | 145 | 0.569940 |
| Sample06 | 87924016 | 12695013324 | 12.7G | 12369373961 | 11751217846 | 0.974349 | 0.925656 | 144 | 144 | 0.598578 |
| Sample07 | 49394180 | 7138495628 | 7.14G | 7031003831 | 6787488038 | 0.984942 | 0.950829 | 144 | 144 | 0.492438 |
| Sample08 | 55500062 | 8024601437 | 8.02G | 7903871021 | 7627315406 | 0.984955 | 0.950491 | 144 | 144 | 0.463274 |
| Sample09 | 60756224 | 8792932412 | 8.79G | 8639370240 | 8304738404 | 0.982536 | 0.944479 | 144 | 144 | 0.464453 |
| Sample10 | 55823930 | 8052677014 | 8.05G | 7881218129 | 7517781014 | 0.978708 | 0.933575 | 144 | 144 | 0.505271 |
| Sample11 | 58793960 | 8488478733 | 8.49G | 8302727633 | 7906894172 | 0.978117 | 0.931485 | 144 | 144 | 0.468602 |
| Sample12 | 60007224 | 8669617727 | 8.67G | 8481076959 | 8079037222 | 0.978253 | 0.931879 | 144 | 144 | 0.466449 |
- sample id: 样本ID
- total reads: 样本序列总数
- total bases: 样本碱基总数
- total giga bases: 样本碱基总数(G为单位)
- q20 bases: 质量分高于20(错误率0.01)的碱基总数
- q30 bases: 质量分高于30(错误率0.001)的碱基总数
- q20 rate: 质量分高于20(错误率0.01)的碱基占比(Q20)
- q30 rate: 质量分高于30(错误率0.001)的碱基占比(Q30)
- read1 mean length: read1平均长度
- read2 mean length: read2平均长度
- gc content: GC含量
3.2 转录本拼接
02.Assemble/
├── Genes.Transcripts
│ ├── NR_genes.fa.gz [非冗余基因序列]
│ └── [样本名]_transcripts.fa.gz [各样本组装得到的转录本]
└── QUAST
├── NRGenes
│ └── report.html [非冗余基因序列的quast质量评估报告,文件夹中其他文件为报告的数据文件,可忽略]
└── SampleTranscripts
└── report.html [各样本组装得到的转录本的quast质量评估报告,文件夹中其他文件为报告的数据文件,可忽略]
对于每一个样品,使用拼接软件Trinity[4] ( https://github.com/trinityrnaseq/trinityrnaseq/wiki/ ) 对预处理后(质控,去宿主, 去rRNA)的clean reads进行从头组装(参数:--min_contig_length 200)得到 每个样本的转录本([样本名]_transcripts.fa.gz); 挑选每个基因最长的转录本序列,作为该基因的序列代表; 然后整合所有样品的基因序列,并使用CD-HIT-EST [5] 去冗余(参数:-c 0.95, 设定序列一致性阈值为 0.95),得到非冗余基因集合(NR_genes.fa.gz)。
转录本拼接
Trinity是一款由 Broad Institute 和 Hebrew University of Jerusalem合作研发的, 针对RNA-seq数据的,高效稳定的转录组拼接软件。 其结合了三个独立的软件模块依次对大量的RNA-seq数据进行了处理拼接,分别为: 茧(Inchworm)、蛹(Chrysalis)、蝶(Butterfly)[3]。主要过程如下:
- 茧(Inchworm):分解reads,构建k-mer(k=25)字典,选择种子k-mer并进行两边延伸,形成contig。
- 蛹(Chrysalis): 将有重叠的contigs聚类,构成components,每个component就成为一组可变剪切isoform或同源基因可能的表征的集合。 每个component会有相应的de Bruijn graph。
- 蝶(Butterfly): 化简每个component的de Bruijn graph,输出可变剪切亚型的全长转录本,并梳理对应于旁系同源基因的转录本, 最终得到拼接结果文件:[样本名]_transcripts.fa.gz。
拼接原理图形展示如下:
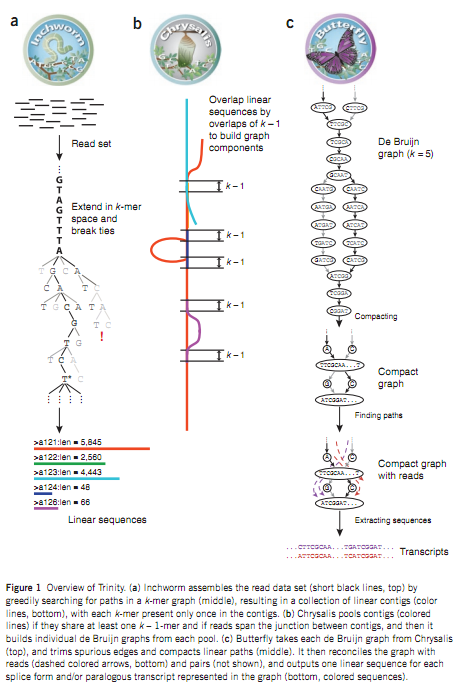
图3-2-1 Trinity拼接原理图
拼接得到的转录本序列信息以FASTA格式([样本名]_transcripts.fa.gz)储存,如下所示:
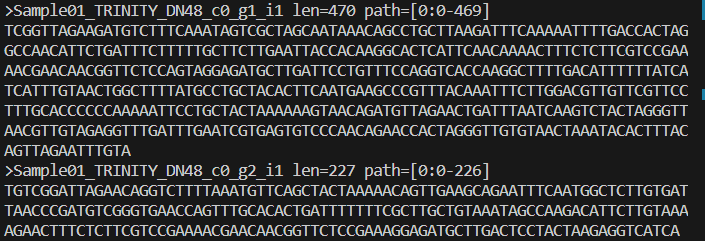
图3-2-2 拼接得到的转录本
其中大于号>后紧跟转录本的id号,len=后面为转录本的长度,即该转录本的碱基数, path为从 de Bruijn Graph subComponent中经历的路径。 其后为该转录本的碱基序列。每个转录本的id号包括多个字段(下划线隔开), 第一个字段为样本ID(如图中的Sample01),最后三个字段为c_g_i: 其中 "c" (如图中的TRINITY_DN48_c0)为拼接过程形成的 de Bruijn Graph Component;g为subcomponet, 可以看作为广泛意义上的gene;i代表转录本isoform(一个基因可以有多个转录本)。 即:c_g为基因ID,c_g_i为c_g基因不同的转录本。 详细解释见Trinity官方网站: https://github.com/trinityrnaseq/trinityrnaseq/wiki
CD-HIT-EST 去冗余
相同环境或者条件下的样品间可能共有很多微生物或基因, 不同基因的丰度在样本间的变化可以反映样本间的共性和差异, 因此需要去冗余构建非冗余基因集(non-redundant gene catalog)来描述所有基因的整体信息。 CD-HIT-EST[5] 去冗余的基本思路是首先对所有序列按照长度进行排序, 然后从最长的序列开始,形成第一个序列cluster,依次对序列进行处理, 如果新的序列与已有的序列cluster的代表序列的相似性在cutoff以上则把该序列加到该序列cluster中, 否则形成新的序列cluster。详情请参考 CD-HIT 官方介绍 。
NR_genes.fa.gz文件保存了CD-HIT-EST软件去冗余后基因序列。
转录本序列信息统计
对于每个样本最终得到的转录本以及去冗余后的基因序列, 我们使用 QUAST[6] 软件进行质量评估。
每个样本最终得到的转录本的QUAST质量评估报告如图3-2-3
图3-2-3 每个样本转录本的quast质量评估报告
图中展示了组装得到的转录本 长度分布情况。N50: reads组装后会获得一些不同长度的转录本。 将所有的转录本按照从长到短进行排序,然后把转录本的 长度按照这个顺序依次相加,当相加的长度达到转录本总长度的一半时, 最后一个加上的转录本长度即为转录本 N50。 鼠标悬浮在相应字段上,能查看每个字段的详细解释。
去冗余后的基因序列的QUAST质量评估报告见结果文件夹, 解读方法类似。
3.3 物种注释和功能注释
03.Annotation/ └── gene_info.xls [非冗余基因物种注释和功能注释结果的汇总表格]
物种注释
- 使用DIAMOND软件[7] 将 非冗余基因(NR_genes.fa.gz) 与从 NCBI 的 NR(Version: 2020-6-25) 数据库中抽提出的细菌(Bacteria)、真菌(Fungi)、古菌(Archaea)和病毒(Viruses) 序列进行比对(参数: blastx --top 3 -e 1e-5 --id 70; 使用 生科云 云流程可调整阈值)。
- 由于每一条序列可能会有多个比对结果,得到多个不同的物种分类信息,为了保证其生物意义, 我们使用BASTA软件[8] (参数: --alen 100 --identity 80 --evalue 0.00001 --minimum 3 --best_hit 1 --maj_perc 60), 基于LCA算法, 将出现第一个分支前的分类级别,作为该序列的物种注释信息。 由于蛋白可能是直系同源蛋白(即在不同物种之间同源的蛋白), LCA算法注释结果可能不太理想(如果不考虑准确性, 只考虑注释结果的多少), 因此, 想要注释到的物种分类水平多一些, 可以在云流程中选择将best hit(最佳匹配序列的物种分类) 作为最终的物种注释结果。
功能注释
我们对五个功能数据库进行了注释: GO,KEGG,CAZy, eggNOG COG和CARD, 它们各自的介绍及注释所用软件如下:
| 数据库 | 简介 | 详细信息 | 软件及参数 |
|---|---|---|---|
| GO | Gene Ontology,一套国际标准化的基因功能描述的分类系统 | GO分为三大类ontology:生物过程(Biological Process)、 分子功能(Molecular Function)和细胞组分(Cellular Component), 分别用来描述基因编码的产物所参与的生物过程、所具有的分子功能及所处的细胞环境。 GO的基本单元是term,每个term有一个唯一的标示符 (由“GO:”加上7个数字组成,例如GO:0072669); 每类ontology的term通过它们之间的联系(is_a, part_of, regulate)构成一个有向无环的拓扑结构。 详见http://www.geneontology.org/。 | emapper(v2.0.1), 参数: -m diamond --translate --seed_ortholog_evalue 0.00001 |
| KEGG | Kyoto Encyclopedia of Genes and Genomes | 系统分析基因产物和化合物在细胞中的代谢途径以及这些基因产物的功能的数据库。 它整合了基因组、化学分子和生化系统等方面的数据,包括代谢通路(KEGG PATHWAY)、 药物(KEGG DRUG)、疾病(KEGG DISEASE)、功能模型(KEGG MODULE)、 基因序列(KEGG GENES)及基因组(KEGG GENOME)等等。 KO(KEGG ORTHOLOG)系统将各个KEGG注释系统联系在一起, KEGG已建立了一套完整KO注释的系统,可完成新测序物种的基因组或转录组的功能注释。 详见 http://www.genome.jp/kegg/ 。 | emapper(v2.0.1), 参数: -m diamond --translate --seed_ortholog_evalue 0.00001 |
| CAZy | Carbohydrate-Active Enzymes Database | 研究碳水化合物酶的专业级数据库,主要涵盖 6 大功能类: 糖苷水解酶(Glycoside Hydrolases ,GHs),糖基转移酶(Glycosyl ransferases,GTs), 多糖裂合酶(Polysaccharide Lyases,PLs),碳水化合物酯酶(Carbohydrate sterases,CEs), 辅助氧化还原酶(Auxiliary Activities , AAs)和 碳水化合物结合模块(Carbohydrate-Binding Modules,CBMs)。 详见:http://www.cazy.org/ | emapper(v2.0.1), 参数: -m diamond --translate --seed_ortholog_evalue 0.00001 |
| eggNOG | evolutionary genealogy of genes: Non-supervised Orthologous Groups | eggNOG V3 涵盖了 1,133 个物种的基因,构建了包含 24 类,约 70 万个 Orthologous Groups。 详见:http://eggnogdb.embl.de/ | emapper(v2.0.1), 参数: -m diamond --translate --seed_ortholog_evalue 0.00001 |
| CARD | Comprehensive Antibiotic Resistance Database | CARD数据库目前使用最广泛的抗性基因数据库,目前包括3997个抗性基因分类, 并在线提供各个分类名称与PDB、NCBI等数据库的搜索接口,方便后续分析。 详见: https://card.mcmaster.ca/ | diamond(v0.8.22), 参数: blastx --top 3 -e 1e-5 --id 70 --more-sensitive |
- 使用eggnog-mapper(emapper)[9] (参数: -m diamond --translate --seed_ortholog_evalue 0.00001)对非冗余基因(NR_genes.fa.gz) 进行功能注释,获得KEGG, GO, EGGNOG COG, CAZy数据库的注释结果。
- 使用DIAMOND软件[7], 将非冗余基因(NR_genes.fa.gz) 与 CARD数据库ARO序列比对(参数: blastx --top 3 -e 1e-5 --id 70 --more-sensitive; 使用 生科云 云流程可调整阈值), 获得CARD数据库的注释结果。
结果文件夹中, gene_info.xls是非冗余基因物种注释和功能注释结果的汇总表格, 其各列解释如下:
- Gene ID: 基因编号
- KEGG Gene ID: KEGG数据库功能注释结果, KO是KEGG通路的基本单元, 用于KEGG通路富集分析
- GO: GO数据库功能注释结果
- STRING Protein: EGGNOG COG数据库功能注释结果, STRING数据库收录了COG的蛋白互作网路, COG是STRING蛋白互作网络物种无关部分的基本单元, 用于差异蛋白互作网络分析
- CAZY: CAZy数据库功能注释结果
- Taxonomy: 物种注释结果
- CARD: CARD数据库功能注释结果
- KEGG description: KEGG数据库KO(KEGG ORTHOLOGY)功能描述
3.4 定量
基因定量
04.Gene/ ├── gene_count.xls [基因fragments count表达量矩阵] ├── gene_deseq2.xls [用DESeq2的方法校正后的表达量矩阵] ├── gene_edger_TMM.xls [用edgeR的方法校正后的表达量矩阵] ├── gene_fpkm.xls [FPKM表达量矩阵] └── gene_tpm.xls [TPM表达量矩阵]
使用RSEM软件[10](参数:--paired-end --bowtie2) 对非冗余基因进行定量,得到基因的fragments count, FPKM, TPM表达量矩阵;
FPKM和TPM的计算请参照文献中的公式,这两个表达量可用于比较样本或分组之间的表达量差异,也能用于比较基因之间的表达量差异
以gene_count.xls为输入,筛除检出率(count不为0的比例)小于0.25的基因, 用R语言DESeq2包[11]和edgeR包[12] 分别计算校正后的表达量gene_deseq2.xls和gene_edger_TMM.xls; 这两个表达量表格可用于比较样本或分组之间的表达量差异,但不能用于比较基因之间的表达量差异。 请注意,这个两个文件并不能作为DESeq2和edgeR的直接输入文件, 也不能用于计算DESeq2和edgeR的FoldChange,DESeq2和edgeR只能用gene_count.xls作为输入文件, 因为它们已经将校正过程编码到模型中
物种定量
05.Taxonomy/ [物种丰度分析结果文件夹] ├── Class.xls [纲水平物种丰度表] ├── Family.xls [科水平物种丰度表] ├── gene_taxonomy.xls [带物种注释的基因丰度表] ├── Genus.xls [属水平物种丰度表] ├── Kingdom.xls [界水平物种丰度表] ├── Order.xls [目水平物种丰度表] ├── Phylum.xls [门水平物种丰度表] └── Species.xls [种水平物种丰度表]
结合基因表达量矩阵(默认gene_tpm.xls,云流程可调整)和基因的物种注释结果(gene_info.xls的Taxonomy列), 加和注释到每个物种水平的基因表达量,得到每个物种水平的丰度表,用于后续可视化分析。
功能定量
06.Function/
└── [功能数据库名称] [功能数据库对应的结果文件夹]
├── Abundance [功能丰度表文件夹]
│ ├── All.[功能数据库名称].[功能分类].xls [功能数据库对应功能分类的丰度表]
│ ├── [功能数据库名称]_abundance_mixed.xls [按物种分类分层的基本功能丰度表]
│ ├── [功能数据库名称]_abundance_unstratified.detail.xls [带功能详细注释的基本功能丰度表]
│ └── [功能数据库名称]_abundance_unstratified.xls [功能数据库对应的基本功能丰度表]
└── GeneCount [基因数目文件夹]
├── All.[功能数据库名称].[功能分类].xls [功能数据库对应功能分类的“成员基因”数目表]
├── [功能数据库名称].detail.xls [带功能详细注释的基本功能“成员基因”数目表]
└── [功能数据库名称].xls [功能数据库对应的基本功能“成员基因”数目表]
1. 基因数目
对于每个功能数据库的基因功能注释结果(gene_info.xls对应数据库的列), 我们统计注释到每个基本功能或者功能分类的基因数目,用于后续可视化分析。 注意区别这里的基因数目和上文说到的fragments count表达量矩阵(gene_count.xls), 这里的基因数目数的是基因的个数, 不属于丰度的范畴, 而fragments count表达量矩阵 数的是每个样本的fragment (read pair, 即一对read)数。
2. 功能丰度
结合基因表达量矩阵(默认gene_tpm.xls,云流程可调整)和基因的功能注释结果(gene_info.xls对应数据库的列), 加和注释到每个基本功能和功能分类的基因表达量,得到各功能数据库(KEGG, GO, EGGNOG COG, CAZy, CARD)的 功能丰度表,用于后续可视化分析。
3.5 基因表达量可视化
3.5.1 挑选差异表达基因
04.Gene/01.DEG/ ├── [分组方案]_[处理组].vs.[对照组] │ ├── [分组方案]_[处理组].vs.[对照组]_All_KEGG.xls [KEGG基因差异分析的完整结果,用于下游KEGG GSEA分析] │ ├── [分组方案]_[处理组].vs.[对照组]_All.xls [基因差异分析的完整结果,用于下游GO GSEA分析] │ ├── [分组方案]_[处理组].vs.[对照组]_DEG_KEGG.xls [显著差异的KEGG基因集,用于下游KEGG ORA分析] │ ├── [分组方案]_[处理组].vs.[对照组]_DEG_STRING.xls [显著差异的STRING基因集,用于下游PPI(蛋白互作网络)分析] │ ├── [分组方案]_[处理组].vs.[对照组]_DEG.xls [显著差异的基因集,用于下游GO ORA分析,Venn|upset图,聚类热图] │ ├── [分组方案]_[处理组].vs.[对照组]_Volcano.svg [差异分析火山图] │ ├── heatmap.svg [差异最显著的基因表达量聚类热图] │ ├── input.xls [聚类热图输入表达量矩阵] │ └── table.xls [聚类热图最终数据] └── VennUpSet ├── DEG_upset.svg [各差异基因集之间的UpSet图] ├── DEG_upset_venn_intersects.xls [各差异基因集之间的交集] ├── DEG_venn.svg [各差异基因集之间的Venn图] ├── summary_up_down.svg [各差异基因集上调,下调数目汇总柱形图] └── summary_up_down.xls [各差异基因集上调,下调数目汇总表]
在宏转录组中,确定某个基因在不同的样品中的表达量是否有差异是分析的核 心内容之一。获得基因表达量后,即可对表达数据进行统计学分析,进而筛选不 同样本之间显著差异的基因。差异分析主要分为四个步骤:
- 以gene_count.xls为输入,筛除检出率(count不为0的比例)小于0.25的基因,这些基因通常不服从分析模型的数据分布假设,因而不适合做差异分析
- 对count进行标准化(normalization),主要是对测序深度的 校正。
- 通过统计学模型进行假设检验,计算p value
- 进行多重假设检验校正(BH方法),获得经校正的p value (结果表中的padj)
针对不同的实验情况,我们选用合适的软件进行基因表达差异显著性分析, 具体如下表所示:
表3-5-1-1 表达差异分析所用软件
| 类型 | R语言包 | 标准化方法 | 数据分布假设 | pvalue计算模型 | padj计算方法 |
|---|---|---|---|---|---|
| 有生物学重复 | DESeq2[11] | DESeq2 | 负二项分布 | 广义线性模型 | BH |
| 无生物学重复 | edgeR[12] | TMM | 负二项分布 | 广义线性模型 | BH |
一般来说,如果一个基因在两组样品中的表达量差异达到两倍以上,我们认 为这样的基因是具有表达差异的。为了判断两个样品之间的表达量差异究竟是由 于各种误差导致的还是本质差异,我们需要对所有基因在这两个样本中的表达量 数据进行假设检验。而宏转录组分析是针对成千上万个基因进行的,这样会导致假 阳性的累积,基因数目越多,假设检验的假阳性累积程度会越高,所以引入 padj 对假设检验的 P-value 进行校正,从而控制假阳性的比例 。
差异基因的筛选标准是非常重要的,我们给出的标准|log 2 (FoldChange)| > 1 & padj< 0.05 是常用的经验值,也是我们分析流程的默认值。在实际项目中可以根据情况灵活选择,例如,差 异倍数可以选择 1.5 倍,也可以选择 3 倍,padj 常用的阈值包括 0.01、0.05、0.1 等。若按照以上标准筛选得到的差异基因过少,很有可能导致后面的功能富集分 析没有显著性结果。若项目实验只关注某几个基因的表达情况(如基因敲除), 不在意富集结果,从下面的差异分析表格中筛选关注的那几个基因即可。反之, 如果得到的差异基因数目过多,不利于后续目标基因的筛选,这个时候可使用更 严格的阈值标准进行筛选。生科云的云流程提供了调整筛选阈值的途径。
结果文件夹中,[分组方案]_[处理组].vs.[对照组]_*.xls表格文件的各字段解释如下
- Features: 基因ID
- log2FoldChange: 处理组与对照组基因表达水平的比值,再经过差异分析软件收缩模型处理,最后以 2 为底取对数
- logCPM: edgeR结果特有,log2(所有样本校正后表达量均值)
- LR: edgeR结果特有,检验统计量,用于计算p值,无需关注
- baseMean: DESeq2结果特有,所有样本校正后表达量均值
- pvalue: 显著性检验的p值
- padj: BH方法校正后的p值
- -log10(pvalue): -log10(pvalue)
- -log10(padj): -log10(padj)
- KEGG Gene ID: KEGG数据库对应的基因ID
- GO: GO数据库对应的基因本体条目
- STRING Protein: STRING数据库对应的蛋白ID
火山图同时展示了差异分析的-log10(pvalue)和log2FoldChange
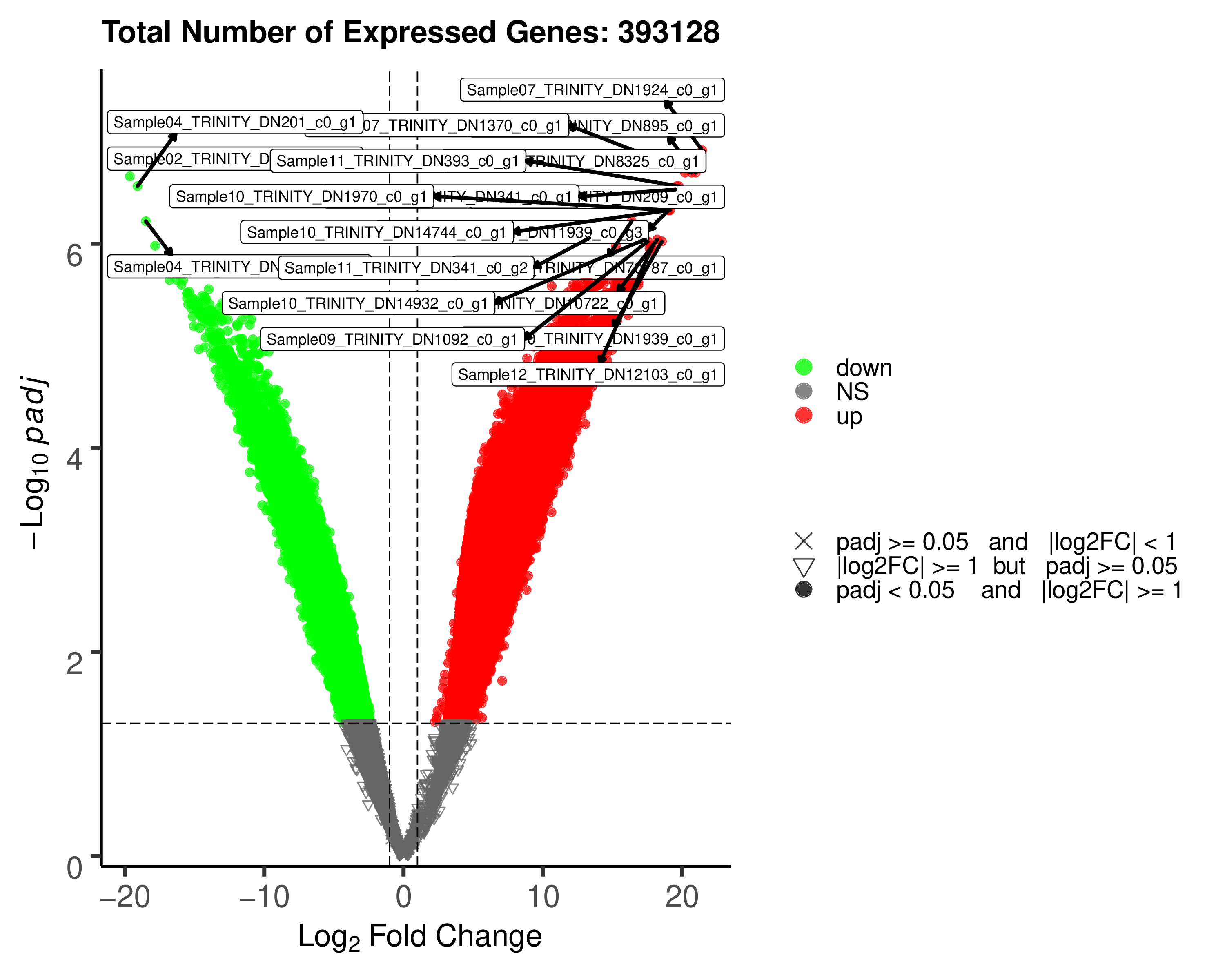
图3-5-1-1 差异分析火山图
标出基因名的是差异显著且padj最小的基因
summary_up_down.svg汇总了各差异基因集上调(处理组表达量高于对照组),下调数目

图3-5-1-2 各差异基因集上调,下调数目汇总柱形图
如果有多个比较方案,多个差异基因集,我们将用Venn图和UpSet图(DEG_venn.svg|图3-5-1-3, DEG_upset.svg|图3-5-1-4)展示各个差异基因集之间的交集大小(共有的差异基因数目),以及各个差异基因集特有的差异基因。Venn图和UpSet图表达的信息是一样的,只是用了不同的数据可视化形式。Venn图最多能展示五个差异基因集的关系,而UpSet图能展示的差异基因集个数能够更多。

图3-5-1-3 各差异基因集之间的Venn图

图3-5-1-4 各差异基因集之间的UpSet图
差异基因聚类热图(heatmap.svg,图3-5-1-5)展示了最显著的(padj最小)的基因的表达量矩阵。为了与p值计算方法保持一致,对于没有生物学重复的比较方案,我们用6.Quant/gene_edger_TMM.xls作为输入,有生物学重复则用6.Quant/gene_deseq2.xls。为了保证图片的可读性,我们展示的基因不会超过20个(云流程可调整)

图3-5-1-5 差异最显著的基因聚类热图
图中用颜色代表表达量,数据跨度太大会导致差异无法区分,我们对表达量进行标准化处理(减去平均值,除以标准差);聚类到一起的样本表达模式比较接近
3.5.2 富集分析
04.Gene/02.Enrich
├── GSEA
│ ├── GO
│ │ └── [分组方案]_[处理组].vs.[对照组]
│ │ ├── circular_cnetplot.svg [富集分析p值(p.adjust)最小的条目的环状基因概念网络]
│ │ ├── cnetplot.svg [富集分析p值(p.adjust)最小的条目的基因概念网络]
│ │ ├── dotplot.svg [富集分析p值(p.adjust)最小的条目的气泡图]
│ │ ├── emapplot.svg [富集分析p值(p.adjust)最小的条目的emapplot]
│ │ ├── enrich.xls [富集分析结果表格]
│ │ ├── heatplot.svg [富集分析p值(p.adjust)最小的条目的基因概念瀑布图]
│ │ ├── summary_gseaplot.svg [富集分析p值(p.adjust)最小的条目的gseaplot]
│ │ └── treeplot.svg [富集分析p值(p.adjust)最小的条目的聚类树图]
│ └── KEGG
│ └── [分组方案]_[处理组].vs.[对照组]
│ ├── circular_cnetplot.svg
│ ├── cnetplot.svg
│ ├── dotplot.svg
│ ├── emapplot.svg
│ ├── enrich.xls
│ ├── heatplot.svg
│ ├── KEGG_pathview [包含富集分析p值(p.adjust)最小的KEGG通路的通路图的文件夹]
│ ├── summary_gseaplot.svg
│ └── treeplot.svg
└── ORA
├── GO
│ └── [分组方案]_[处理组].vs.[对照组]
│ ├── circular_cnetplot.svg
│ ├── cnetplot.svg
│ ├── dotplot.svg
│ ├── emapplot.svg
│ ├── enrich.xls
│ ├── heatplot.svg
│ └── treeplot.svg
└── KEGG
└── [分组方案]_[处理组].vs.[对照组]
├── circular_cnetplot.svg
├── cnetplot.svg
├── dotplot.svg
├── emapplot.svg
├── enrich.xls
├── heatplot.svg
├── KEGG_pathview
└── treeplot.svg
注: 同名文件解释相同,不赘述
本报告所有富集分析使用的都是R语言clusterProfiler包[13]。
关于本报告中使用的富集分析的详细介绍,请参考clusterProfiler包教程
为了保证结果图的可读性,cnetplot.svg, heatplot.svg, gseaplot.svg图选取最显著的(p.adjust最小)5个条目作图。dotplot.svg, treeplot.svg, emapplot.svg图选取最显著的20个条目作图。云流程可自由调整
ORA分析
在上游分析得到了组间表达量有显著性差异的基因数据后,我们可以在基因注释文件中查到单个基因的功能。此外,我们还可以探究这些具有研究意义的基因中,是否显著地对应着一些功能分类的基因的集合,从而推断差异表达基因主要参与的生物学过程
KEGG代谢通路数据库中,有很多前人归纳的生物的代谢通路,一个代谢通路包含了一定数量的基因,那么我们得到的差异表达的基因集合中,会不会存在一种情况,即大量的基因集中出现在一个或几个代谢通路中,如果是这样,那么不同处理所影响的生物学过程就更加明确了。类似的,常见的功能富集分析还有GO条目富集分析。
ORA分析结果表格(ORA/[KEGG|GO]/enrich.xls)各列含义如下
- ID: 富集到的条目(如KEGG通路)的ID
- Description: 富集到的条目(如KEGG通路)的描述
- GeneRatio: 匹配到该条目的差异基因数/差异基因总数
- BgRatio: 该条目一共有多少基因/所有条目一共有多少基因
- pvalue: 显著性检验p值
- p.adjust: BH方法校正后的p值
- qvalue: 校正错误发现率(FDR)的p值
- geneID: 与该条目相匹配的差异基因ID或基因名
- Count: 匹配到该条目的差异基因数
围绕该结果表格,我们做了很多可视化。
GO ORA

图3-5-2-1 富集分析p值(p.adjust)最小的条目的气泡图(dotplot.svg)
点大小对应Count: 匹配到该条目的差异基因数,即enrich.xls表Count列(ORA结果)或者core_enrichment列基因个数(GSEA结果);颜色对应p.adjust; BH方法校正后的p值; 横坐标对应GeneRatio: 匹配到该条目的差异基因数/差异基因总数

图3-5-2-2 富集分析p值(p.adjust)最小的条目的基因概念网(cnetplot.svg)
图中,每一簇的节点的中心是GO条目,这些外围的节点是对应的基因(即enrich.xls表geneID列(ORA分析)或者core_enrichment列(GSEA分析)),有一些基因可能会同时拥有多种GO条目的功能描述。 图中右侧的图例,size表示某一个GO条目对应有多少个基因(即enrich.xls表Count列(ORA结果)或者core_enrichment列基因个数(GSEA结果)),在作图区中表现为,一个GO条目对应的基因个数越多,则该GO条目的圆点越大;而所有的基因节点的面积大小是相同的,这些节点没有表征个数多少的含义; 图中右侧的图例,fold change,它指的是上游的差异分析中,得到的在某个比较组合中每一个基因的log2FoldChange值,作图区中的基因的节点的颜色跟log2FoldChange值是直接对应的;而中心的GO条目圆点的颜色都是相同的,它们的颜色不表征log2FoldChange值的含义; 给线条涂了不同的颜色,使得不同的GO条目延伸出来的线条颜色是不同的;

图3-5-2-3 络富集分析p值(p.adjust)最小的条目的环状基因概念网络(circular_cnetplot.svg)
circular_cnetplot.svg是前面的cnetplot.svg的一种特殊画法,图中元素的内容本质上是一样的, 只是把所有的基因节点与GO条目圆点都尽量画在一个圆周上。

图3-5-2-4 富集分析p值(p.adjust)最小的条目的基因概念瀑布图(heatplot.svg)
这个热图是非聚类热图,表达的意思和cnetplot.svg相似。横轴代表基因,按照英文字母从A到Z的顺序,把基因从左到右排列;纵轴代表GO条目,按照英文字母从A到Z的顺序,把GO条目从下往上排列。 图中右侧的图例,fold change,它指的是上游的差异分析中,得到的在某个比较组合中每一个基因的log2FoldChange值,作图区中的基因对应的单元格的颜色跟log2FoldChange值是有关联的。由于log2FoldChange只跟基因有关,而与GO条目无关,所以在横轴的某一个基因的上方,如果出现了多个有颜色的单元格,那么它们的颜色应该是相同的;而如果在某个位置没有出现涂色的单元格,则表示enrich.xls表中在纵轴坐标对应的GO条目的geneID列(ORA分析)或者core_enrichment列(GSEA分析)中不包含横轴对应的这个基因。
KEGG ORA
除上文介绍的富集结果图外,KEGG ORA在KEGG_pathview文件夹中还有最显著的通路图。
图3-5-2-5 富集分析p值(p.adjust)最小的KEGG通路的通路图
图中红 色表示上调基因,绿色表示下调基因。鼠标悬停于标记节点,可弹出差异基因的 细节信息,主要包括基因 ID, log2FoldChange和 padj 值。 点击各个节点,可以连接到 KEGG 官方数据库中各个节点的具体信息页。
GSEA分析
ORA分析关注的是差异表达基因,然而可能筛不到差异表达基因。GSEA可以用所有基因参与分析,GSEA在一个基因集(如某个GO条目或者KEGG通路)中累加计算一个统计量,因此可以用于检测一些协同作用的小效应,这很重要,因为很多表型差异是由很多协同作用的小效应造成的。
给定一个预先定义的基因集S(如一个GO条目的成员基因集),和一个依据某个指标排好序的基因列表L(本报告中L采用所有基因的log2FoldChange排序),GSEA的目的在于检验S的成员是随机分散在L中(p.adjust>0.05),还是主要集中在L的底部或者顶部(p.adjust<0.05); 在本报告中,enrich.xls中的p.adjust<0.05也就意味着对应富集条目(如某个GO条目或者KEGG通路)的大部分成员基因低表达,或者高表达
除上文介绍的富集结果图外,GSEA分析还有一个专门为其量身打造的GSEA图
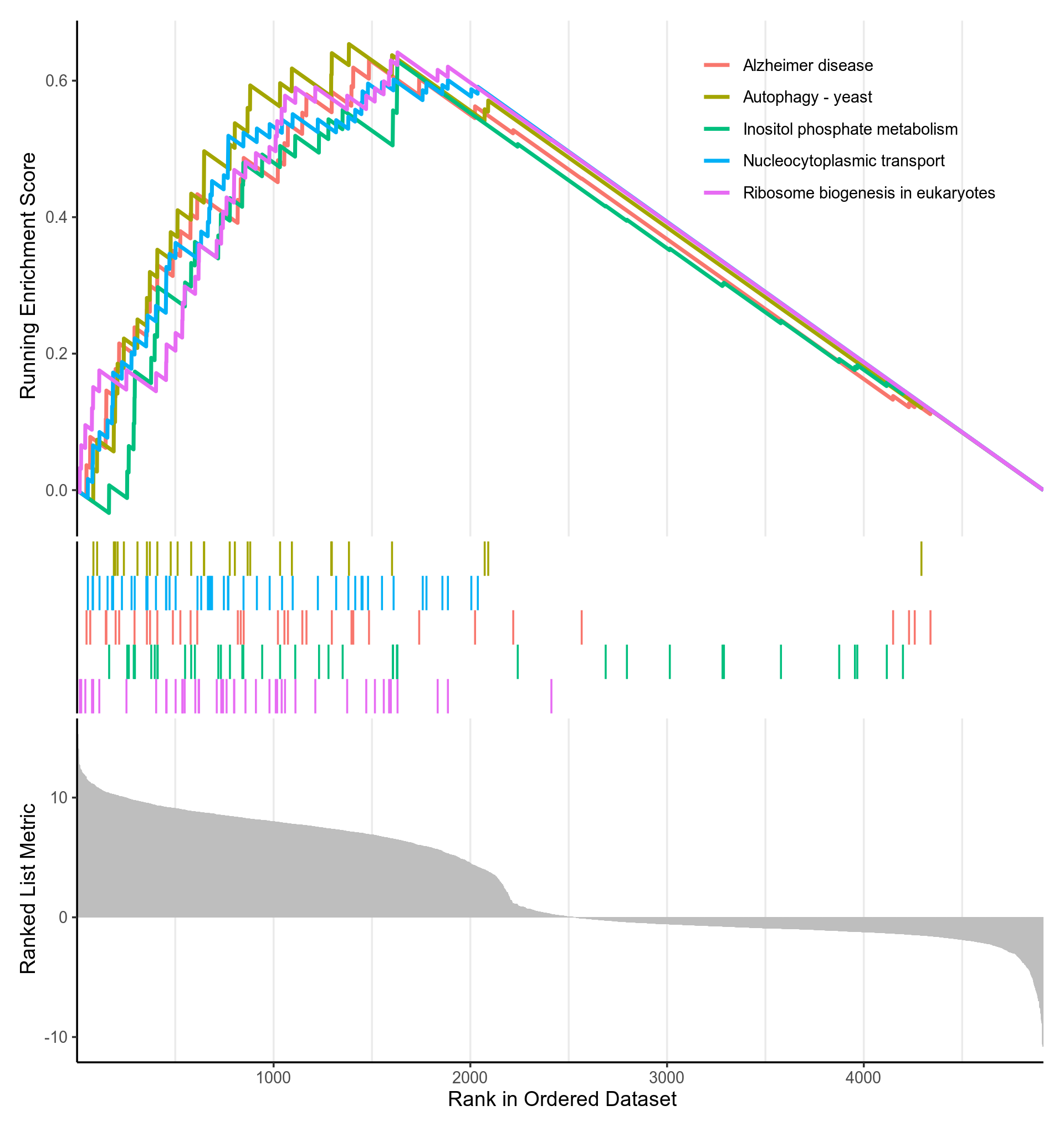
图3-5-2-6 GSEA富集分析p值(p.adjust)最小的KEGG通路的gseaplot
- 该图分为三部分。横坐标都代表排好序的基因列表L,如1代表log2FoldChange最高的基因。
- 对于上部分:纵坐标Running Enrichment Score的计算方式如下:从基因集L的第一个基因开始,计算一个累计统计值。当遇到一个落在S里面的基因,则增加统计值。遇到一个不在S里面的基因,则降低统计值。如果S的成员是随机分散在L中,则对应基因集的线(某个KEGG通路的线,一种颜色代表一个KEGG通路)应该是平缓的。如果存在一个单峰,则表示大部分S的成员聚集在峰的区域(不包含峰后面的部分),该区域的基因即enrich.xls的core_enrichment列(S的子集),该单峰的纵坐标即为富集得分(enrich.xls的enrichmentScore),横坐标即为enrich.xls的rank列
- 中间部分用竖线代表S的成员(某个KEGG通路的成员),有竖线,代表S包含该基因(KEGG通路包含该基因),竖线密集的地方就是Running Enrichment Score峰值出现的部位,每种颜色代表一个单独的S(KEGG通路)
- 下部分展示了排好序的基因列表L的排序指标,这里的纵坐标就是log2FoldChange
GSEA分析的enrich.xls的各列解释如下:
- ID: 富集到的条目(如KEGG通路)的ID
- Description: 富集到的条目(如KEGG通路)的描述
- setSize: 富集到的条目(如KEGG通路)的所有成员基因个数
- enrichmentScore: 富集分数
- NES: 标准化富集分数,GSEA的关键统计量,用于计算p值(置换检验)
- pvalue: 显著性检验p值(置换检验)
- p.adjust: 校正后p值(BH方法)
- qvalues: 校正后p值(FDR方法)
- rank: Running Enrichment Score达到最高值的排序位置(图3-5-2-6对应峰值的横坐标)
- leading_edge: 这些指标是用来表征core_enrichment(S的子基因集)的算法度量,无需关注。实在好奇,请参考GSEA官方解释
- core_enrichment: 核心富集基因集,即enrichmentScore峰值往前的S的子基因集
语义相似性分析
富集结果中的emapplot.svg和treeplot.svg是富集条目之间语义相似性分析结果图,这里使用的相似性指数是Jaccard correlation coefficient,计算公式如下:
即两个富集条目所包含基因集合[enrich.xls表geneID列(ORA分析)或者core_enrichment列(GSEA分析)]的交集基因数除以并集基因数

图3-5-2-7 富集分析p值(p.adjust)最小的条目的emapplot(emapplot.svg)
- 节点的大小对应enrich.xls的Count列(ORA结果)或者core_enrichment列基因个数(GSEA结果)
- 节点的颜色对应enrich.xls的p.adjust列
- 节点之间的连线代表Jaccard correlation coefficient。可理解为连线越粗,两个条目共有的基因个数(标准化后)越多

图3-5-2-8 富集分析p值(p.adjust)最小的条目的聚类树图(treeplot.svg)
- 节点的大小对应enrich.xls的Count列(ORA结果)或者core_enrichment列基因个数(GSEA结果)
- 节点的颜色对应enrich.xls的p.adjust列
- 这是一个依据条目之间Jaccard correlation coefficient进行聚类的树图,聚到一起的条目之间的Jaccard correlation coefficient较大,即共有基因数较多。每种颜色代表一个聚类,从聚类中随机挑选一个单词(通常是聚类中词频比较高的单词)作为该聚类的名称写在右边。
3.5.3 蛋白互作网络分析
04.Gene/03.PPI/ ├── [分组方案]_[处理组].vs.[对照组] │ ├── edges.xls [网络边(连线)数据] │ ├── network.html [差异基因的蛋白互作网络图,可动态调整网络] │ └── nodes.xls [网络节点(蛋白)数据]
该分析主要是从STRING数据库[14]搜索差异表达基因对应蛋白的互作网络,用网络图展示。
为了保证网络的可读性,我们对网络做如下筛选
- 初步筛选可信度(combined_score)最高的前1000个互作关系
- 最终筛选连接度(degree)最高的5个节点(关键基因|蛋白),以及与之互作的节点
这些筛选阈值可在云流程调整
图3-5-3-1 差异基因的蛋白互作网络图
为了提升交互体验,可打开一个新的网页操作网络
网络边(连线)数据(edges.xls)各列解释如下:
- protein1: 某个互作关系的第一个蛋白ID
- protein2: 某个互作关系的第二个蛋白ID
- database: 数据库注释的蛋白互作可信度
- database_transferred: 从其他物种(依据同源性)推断的database
- experiments: 实验验证的蛋白互作可信度
- experiments_transferred: 从其他物种(依据同源性)推断的experiments
- coexpression: 共表达可信度
- coexpression_transferred: 从其他物种(依据同源性)推断的共表达可信度
- textmining: 文献报道的蛋白互作可信度
- textmining_transferred: 从其他物种(依据同源性)推断的textmining
- fusion: 存在基因融合的可信度
- neighborhood: 染色体上距离较近的可信度
- neighborhood_transferred: 从其他物种(依据同源性)推断的neighborhood
- cooccurence: 共现性可信度
- combined_score: 综合计算后蛋白互作可信度
网络节点(蛋白)数据(nodes.xls)各列解释如下:
- nodes: 互作的节点ID,对应edges.xls的protein1和protein2
- Gene ID: 基因ID
- Gene Symbol: 基因名
- preferred_name: STRING数据库提供的节点名
- protein_size: 蛋白分子量
- annotation: 蛋白描述
- log2FoldChange: 差异分析中的log2FoldChange
- -log10(padj): 差异分析中的-log10(padj)
- up_down: 差异分析中是上调(up)还是下调(down)
- absolute(log2FoldChange): log2FoldChange绝对值
3.6 物种丰度可视化
05.Taxonomy/ ├── 01.BasicStatistics [基础统计文件夹] │ ├── Barplots [百分比堆积柱形图文件夹] │ │ └── [分组方案名] │ │ └── [分类水平或方式] │ │ ├── barplot.svg [百分比堆积柱形图] │ │ └── table.xls [百分比堆积柱形图数据] │ └── Heatmaps [丰度热图文件夹] │ └── [分组方案名] │ └── [分类水平或方式] │ ├── heatmap.svg [丰度热图] │ └── table.xls [丰度热图数据] ├── 02.GroupComparison [差异比较文件夹] │ ├── ANOVA_Duncan [ANOVA和Duncan检验结果文件夹] │ │ └── [分组方案名] │ │ └── [分类水平] │ │ ├── [分组方案名]_all_feature_anova_results.xls [ANOVA检验结果] │ │ ├── [分组方案名]_all_significant_feature_duncan_results.xls [Duncan检验结果, ANOVA无显著可缺失] │ │ ├── [分组方案名]_all_significant_feature_barplot_of_duncan.svg [Duncan检验结果图, ANOVA无显著可缺失] │ │ └── final_data_for_anova.xls [检验数据(原始丰度log转化,标准化之后)] │ ├── DunnTest [Kruskal-Wallis和Dunn’s Test检验结果文件夹] │ │ └── [分组方案名] │ │ └── [分类水平] │ │ └── DunnTest.xls [Kruskal-Wallis和Dunn’s Test检验结果表格] │ └── LEfSe [LEfSe分析结果文件夹] │ └── [分组方案名] │ └── [分类水平] │ ├── data_for_lefse.xls [LEfSe分析输入文件] │ ├── lefse.cladogram.svg [LEfSe分析结果cladogram图] │ ├── lefse.LDA.xls [LEfSe分析结果表格] │ ├── lefse.normalized.txt [LEfSe标准化后的数据,中间文件] │ └── lefse.svg [LEfSe分析结果LDA图] ├── 03.Correlation [相关性分析结果文件夹] │ ├── CorrelationHeatmap [相关性热图结果文件夹] │ │ └── [分类水平] │ │ ├── Correlation_heatmap.svg [相关性热图] │ │ ├── p_value_matrix.xls [相关系数p值矩阵] │ │ └── spearman_rank_correlation_matrix.xls [spearman相关系数矩阵] │ └── RDA [CCA/RDA结果文件夹] │ └── [分组方案名] │ └── [分类水平] │ ├── [分组方案名]_RDA.envfit.xls [RDA各环境因子坐标,解释方差,p值] │ ├── [分组方案名]_RDA.Factors.PERMANOVA.xls [所有环境因子对微生物群落变异的解释方差的p值计算结果] │ ├── [分组方案名]_RDA_features_location_plot.svg [RDA微生物点图] │ ├── [分组方案名]_RDA.features.xls [RDA微生物点坐标] │ ├── [分组方案名]_RDA.Group.PERMANOVA.xls [分组方式对微生物群落变异的解释方差的p值计算结果] │ ├── [分组方案名]_RDA_sample_location_plot.svg [RDA样本点图] │ ├── [分组方案名]_RDA_sample_location_plot_with_labels.svg [RDA样本点图(带样本名)] │ ├── [分组方案名]_RDA.sample.xls [RDA样本点坐标] │ └── DCA_output.xls [DCA分析各排序轴值] ├── 04.Diversity [多样性分析结果文件夹] │ ├── Alpha [α多样性分析结果文件夹] │ │ └── [分组方案名] │ │ ├── 1-AlphaDiversitySummary │ │ │ ├── alpha-summary.xls [α多样性指数表格] │ │ │ ├── [分组方案名]_alpha_diversity_shannon_entropy.boxplot.svg [shannon指数箱线图] │ │ │ └── [分组方案名]_alpha_diversity_simpson.boxplot.svg [simpson指数箱线图] │ │ ├── 2-AlphaRarefaction [α多样性稀释性曲线] │ │ │ ├── alpha-rarefaction-ggplot2 [ggplot2包画的稀释性曲线拟合曲线文件夹] │ │ │ │ ├── group_mean_observed_features_rarefaction_curve.svg [observed features(contig数)分组均值稀释性曲线拟合曲线] │ │ │ │ ├── group_mean_shannon_rarefaction_curve.svg [shannon指数分组均值稀释性曲线拟合曲线] │ │ │ │ ├── observed_features_rarefaction_curve.svg [observed features(contig数)稀释性曲线拟合曲线] │ │ │ │ └── shannon_rarefaction_curve.svg [shannon指数稀释性曲线拟合曲线] │ │ │ └── alpha-rarefaction-Qiime2 [qiime2画的稀释性曲线文件夹] │ │ │ └── Summary_请点此文件查看.html [qiime2画的稀释性曲线, 文件夹内其它文件为网页数据,可忽略] │ │ └── 3-SignificanceAnalysis [α多样性组间差异比较文件夹] │ │ ├── 1-Wilcox_Test │ │ │ ├── alpha_Category_wilcox_compare_results.xls [Wilcox Test结果表] │ │ │ ├── shannon_entropy_Category_wilcox_compare_boxplot.svg [shannon指数Wilcox Test结果图] │ │ │ └── simpson_Category_wilcox_compare_boxplot.svg [simpson指数Wilcox Test结果图] │ │ └── 2-Kruskal_Wallis │ │ ├── shannon-group-significance │ │ │ └── Summary_请点此文件查看.html [shannon指数Kruskal Wallis检验报告, 文件夹内其它文件为网页数据,可忽略] │ │ └── simpson-group-significance │ │ └── Summary_请点此文件查看.html [simpson指数Kruskal Wallis检验报告, 文件夹内其它文件为网页数据,可忽略] │ └── Beta [β多样性分析结果文件夹] │ ├── NMDS │ │ └── [分组方案名] │ │ ├── bray_curtis_distance_matrix.xls [Bray-Curtis距离矩阵] │ │ ├── nmds_points_ordinates.xls [NMDS分析样本点坐标] │ │ └── NMDS.svg [NMDS分析结果图] │ ├── PCA │ │ └── [分组方案名] │ │ ├── PCA_3D.svg [三维PCA图] │ │ ├── pca_points_ordinates.xls [PCA分析样本点坐标] │ │ └── PCA.svg [二维PCA图] │ └── PCoA │ └── [分组方案名] │ ├── bray_curtis_distance_matrix.xls [Bray-Curtis距离矩阵] │ ├── PCoA_3D.svg [三维PCoA图] │ ├── pcoa_points_ordinates.xls [PCoA分析样本点坐标] │ └── PCoA.svg [二维PCoA图] └── [微生物分类水平].xls
基础统计
百分比堆积柱形图
百分比堆积柱形图用于展示各样本中,每个微生物类别的丰度占比。

图3-6-1 科水平微生物百分比堆积柱形图
当微生物类别超过20个时,只展示占比最高的前20个
丰度热图
丰度热图用于展示各样本中,每个微生物类别的丰度。为了方便比较, 热图中展示的丰度是标准化后的Z-Score,即丰度减去平均值,除以标准差。

图3-6-2 科水平微生物丰度热图
当微生物类别超过20个时,只展示丰度最高的前20个
差异比较
LEfSe
LEfSe[15] (Linear discriminant analysis Effect Size,线性判别分析)即 LDA Effect Size 分析, 是一种发现和解释高纬度数据生物标识(分类单元、通路、基因)的分析工具,可以实现两 个或者多个分组之间的比较,同时也可进行分组内部亚组之间的比较分析。该分析首先使用非参数 Kruskal-Wallis 秩和 检测不同分组间丰度差异显著的微生物 ,然后使用 Wilcoxon 秩和检验上一步的差异微生物 在不同组间子分组中的差异一致性,最后采用线性回归分析(LDA)来估算每个微生物 的丰度对差异效果影响的大小。 LEfSe寻找每一个分组的特征微生物(默认为LDA>2的微生物), 也就是相对于其他分组,在这个组中丰度较高的微生物。 需要注意的是,LEfSe内部会对丰度进行标准化(主要计算每个样本参与分析微生物的丰度比例), 特征微生物原始丰度(TPM)少数情况下不是最高的。

图3-6-3 种水平微生物LEfSe分析LDA柱形图
每一横向柱形体代表一个物种,柱形体的长度对应LDA值,LDA值越高则差异越大。 柱形的颜色对应该物种是哪个分组的特征微生物(在对应分组中的丰度相对较高)。

图3-6-4 种水平微生物LEfSe分析cladogram图
cladogram图由内到外,分别对应界门纲目科属种不同的分类层级,层级间连线代表所属关系。 每个圆圈节点代表一个物种,节点为黄色代表在分组间差异不显著, 不为黄色则代表该物种是对应颜色分组的特征微生物(在该分组丰度显著较高)。 有颜色的扇形区域标注了特征微生物的下属分类区间。
lefse.LDA.xls表格文件从左到右各列解释为:
- feature标识
- 所有样本标准化(算比例)以及log转化后的丰度均值
- 特征feature对应分组,如果为空,表示该feature不是特征feature
- LDA值
- Kruskal-Wallis 秩和检验p值
Kruskal-Wallis和Dunn’s Test检验
使用R语言dunn.test包[16]进行Kruskal-Wallis和Dunn’s Test检验。 Kruskal-Wallis和Dunn’s Test都是基于秩次的非参数检验,对数据分布没有要求。 Kruskal-Wallis 秩和检验通常只能检验一个分组方案所有分组的整体差异, p<0.05只能推断至少有两个分组不一样,如果分组个数大于2, 那具体哪两个分组不一样是无法推断的。这时候我们需要结合Dunn’s Test来进行 多重比较,进而推断具体哪两个分组不一样。
Dunn’s Test的结果表格(DunnTest.xls)各列解释如下:
- Feature: feature标识
- KW_pvalue: Kruskal-Wallis秩和检验p值
- DunnTest_comparison: 分组对
- DunnTest_Z: 分组对Dunn’s Test检验统计量
- DunnTest_PValue: 分组对Dunn’s Test检验p值
- DunnTest_PValueBonferroniAdjusted: 分组对Dunn’s Test检验Bonferroni校正后的p值
ANOVA和Duncan检验
使用R语言ggpubr包[17]进行ANOVA检验, 使用R语言agricolae包[18]进行Duncan检验。 ANOVA和Duncan的关系类似Kruskal-Wallis和Dunn’s Test的关系。区别是,ANOVA和Duncan是有参的检验方法。 要求数据服从正态分布。我们在做该分析之前,筛除检出率(丰度大于0的频率)小于0.2的 features, 并对丰度进行log转化以及标准化(减去平均值,除以标准差),使得数据尽量接近正态分布。
*_all_feature_anova_results.xls为ANOVA检验结果,各列解释如下:
- variable: features标识
- .y.: 无意义
- p: p值
- p.adj: holm方法校正后的p值
- p.format: 保留三位小数的p值
- p.signif: 显著性
- method: 检验使用的方法
*_all_significant_feature_duncan_results.xls为Duncan检验结果,各列解释如下:
- Feature: features标识
- Group: 分组名
- Mean: 分组均值
- SE: 分组标准误
- Duncan significance: 用字母表示的显著性,同一个feature,字母相同的分组差异不显著,不同则显著

图3-6-5 ANOVA和Duncan检验得到的所有显著差异的微生物
横坐标为微生物名字;对于每个微生物,用不同颜色表示不同的分组,两个分组上方如果有相同的字母,则代表差异不显著,否则差异显著。
相关性分析
相关性热图
如果提供了环境因子的数据,比如 pH值、温度值、临床因子等,可以进行相关性热图分析
相关性热图的绘制主要运用了R语言pheatmap包[19]。 相关性热图可以用于分析环境因子或其他临床表型数据与微生物群落或物种之间是否显著相关, 然后计算环境因子与微生物物种间的Spearman相关系数,并用热图展示。 该分析可以挑选出与某种环境因子或疾病显著相关的物种。

图3-6-6 微生物与表型之间的相关性热图
X轴上为环境因子,Y轴为物种。通过计算获得R值(秩相关)和p值。 R值在图中以不同颜色展示,p值若小于0.05则用 * 号标出, 右侧图例是不同R值的颜色区间,同时,左边的色条标注了物种所属门分类。 * 0.01≤ p <0.05,** 0.001≤p < 0.01,*** p < 0.001。 先依据显著的环境因子个数排名(越多越靠前),再依据显著的p值平均值排名(越低越靠前), 该图只展示排名前20个微生物,如果没有显著相关的微生物,则无法作图。
RDA/CCA分析
CCA/RDA的分析主要依赖R语言vegan包[20], 以及用ggplot2[21]进行可视化。 CCA/RDA(DCA判断用哪一种分析)分析是基于对应分析发展的一种排序方法, 将对应分析与多元回归分析相结合,每一步计算均与环境因子进行回归,又称多元直接梯度分析。 RDA是基于线性模型,CCA是基于单峰模型(图7-1)。本报告先进行DCA分析,看最大轴的值是否大于4,如果大于4.0, 就选CCA,否则选RDA。该分析主要用来反映微生物群落与环境因子之间的关系, 可以检测环境因子、样品、微生物群落(或功能)三者之间的关系或者两两之间的关系, 可得到影响样品分布的重要环境驱动因子。该分析给出的所有p值都是反映解释变量 (连续的环境因子,或者分组方案)对微生物群落变异的解释程度是否显著 (简单的说就是解释变量对微生物群落是否有影响,影响是否显著),所有p值都是用R语言vegan包里的置换检验得出的 (permutation test),*_features_location_plot图中的p值 反映了所有连续的数值变量(环境因子)对微生物差异的解释程度(总的p值), 表格*_RDA.envfit中的p值反映了每个环境因子对微生物差异的解释程度, *RDA_sample_location_plot图中的p值反映了分组对微生物差异的解释程度,p<0.05, 解释方差显著。

图3-6-7 种水平微生物CCA/RDA排序图
在CCA/RDA物种排序图内,环境因子用箭头表示, 箭头连线的长度代表某个环境因子与群落分布和种类分布间相关程度的大小(解释方差的大小), 箭头越长,说明相关性越大,反之越小。箭头连线和排序轴的夹角代表某个环境因子与排序轴的相关性大小, 夹角越小,相关性越高;反之越低。环境因子之间的夹角为锐角时表示两个环境因子之间呈正相关关系, 钝角时呈负相关关系。每个点代表一个物种,点越大,对应物种丰度越高, 灰色点代表丰度较低的物种,未在图中注释物种名称,将物种投影到各个环境因子, 对应的值即为该物种倾向于存在的环境(喜欢的环境)。或者说,将物种点与原点连线, 物种间,物种与环境因子间,环境因子间的夹角的余弦值近似于相关系数,至于有多近似, 就要看RDA1/CCA1和RDA2/CCA2两个坐标轴的解释方差百分比有多大,越大越近似。 对于样本排序图,样本点之间的距离近似于微生物群落结构差异程度, 样本点投影到环境因子对应的值近似于该样本真实的环境因子值。

图3-6-8 CCA/RDA样本排序图
环境因子用箭头表示, 箭头连线的长度代表某个环境因子与群落分布和种类分布间相关程度的大小(解释方差的大小), 箭头越长,说明相关性越大,反之越小。箭头连线和排序轴的夹角代表某个环境因子与排序轴的相关性大小, 夹角越小,相关性越高;反之越低。环境因子之间的夹角为锐角时表示两个环境因子之间呈正相关关系, 钝角时呈负相关关系。样本点之间的距离近似于微生物群落结构差异程度, 样本点投影到环境因子对应的值近似于该样本真实的环境因子值。
*_RDA.envfit.xls表格各列解释如下:
- 环境因子
- RDA1: 表征环境因子的方向,与横轴正方向夹角的余弦值
- RDA2: 表征环境因子的方向,与横轴正方向夹角的正弦值
- r2: 环境因子对微生物群落变异的解释方差
- p value: 置换检验计算的解释方差的p值,小于0.05,可推断该环境因子对微生物群落影响显著
*_RDA.*.PERMANOVA.xls表格为所有环境因子(或分组方式)对微生物群落结构变异的解释方差的p值计算结果, 用的是PERMANOVA置换检验的方法,各列解释如下:
- 方差组分(模型部分,剩余部分)
- Df: 自由度
- Variance: 方差剖分
- F: PERMANOVA检验统计量
- Pr(>F): PERMANOVA检验p值
多样性分析
α多样性分析
α 多样性(α diversity)是对单个样品中物种多样性的分析,包含物种组成的丰 富度(species richness)和均匀度(species evenness)两个因素。 shannon (shannon_entropy)和simpson 指数可综合考虑群 落中物种的丰富度和均匀度。群落中有越多的物种(丰富度越高),各物种具有越大的均匀度, 则群落具有越大的α 多样性, shannon和simpson 指数也越高。
本分析使用qiime2[22]软件, 基于样品种水平的物种丰度表(云流程可调整), 计算shannon和simpson 指数。

图3-6-9 shannon指数的箱型图
不同颜色代表不同的分组。
Alpha多样性稀释曲线(Rarefaction Curve)是从样品中随机抽取一定测序量的数据(序列数标准化为TPM,测序量为一百万), 统计它们所属物种数目(unique.contig数目)或多样性指数,以测序数据量与物种多样性来构建的曲线, 以用来说明样品的测序数据量是否合理,并间接反映样品中物种的丰富程度。 曲线的平缓程度反映了测序深度对于观测样本多样性的影响大小,曲线越平缓, 表明测序结果已足够反映当前样本所包含的多样性,继续增加测序深度已无法检测到大量的尚未发现的新物种; 反之,则表明多样性尚未接近饱和,继续增加测序深度将有助于观测到更多的新物种。
图3-6-10 各α多样性指数的稀释性曲线
通过选择下拉框选择不同的α多样性指数或者分组情况(网页上半部分图), 图中X轴代表抽取的序列测序量,Y轴代表相应的α多样性指数值。 网页下半部分图代表在相应抽取测序量时,各分组中仍包含样品数量, 用于查看在一定抽取测序量之下稀释曲线的分组分析是否仍然包括该分组中全部的样本。
图3-6-11是用R语言ggplot2[21] 包画的α多样性指数稀释性曲线的拟合曲线(公式为y ~ log(x)), 这个曲线比较光滑,但也和真实数据存在一定偏差。

图3-6-11 shannon指数稀释性曲线拟合曲线
在得到整体的α多样性指数之后,接下来结合分组信息来用Kruskal Wallis方法 比较在各个样品分组之间α多样性指数是否有显著性差异。 除了使用qiime2推荐的Kruskal Wallis方法,分析还包括使用Wilcox Test 去精确比较了各个组间的显著性差异并做图,图3-6-12显示了shannon指数的组间多重比较结果。

图3-6-12 shannon指数的组间Wilcox多重比较
X轴表示分组名称,Y轴表示α多样性指数。*,**,***分别代表p<0.05, p<0.01, p<0.001。
β多样性分析
β 多样性(β Diversity)即样品间的生物多样性的比较,是对不同样品间的微生物 群落构成进行比较。样本间物种的丰度分布差异程度可通过统计学中的距离进行量化分析, 使用统计算法计算两两样本间距离,获得距离矩阵,可用于后续进一步的 β 多样性分析 和可视化统计分析。 Bray-Curtis 距离是系统聚类法中使用最普遍的一个距离指标,基于物种 的丰度信息计算,是生态学上反映群落之间结构差异性常用的指标之一。 使用R语言vegan[20]包,根据 样品种水平的物种丰度表(云流程可调整)计算 Bray-Curtis 距离, 来评估不同样品间的微生物群落结构差异。
PCA(Principal Component Analysis,主成分分析)是一种传统的数据降维方法,常用于 简化数据集。它运用方差分解,寻找造成样本间差异的主成分(特征值)及其贡献率,选取 两个或三个贡献率最大的主成分进行作图,将多组数据的差异反映在二维或三维坐标图上。

图3-6-13 PCA 分析结果
- 一个点代表一个样本,点与点空间距离近似物种组成结构的差异程度(欧式距离);
- 主成分 1(PC1)和主成分 2(PC2)是造成样品间差异的第一和第二大成分,百分比表示两个主成分对样品差异(方差)的 贡献率。
PCoA(Principal Co-ordinates Analysis,主坐标分析)与 PCA 类似,都是通过降维找出影 响样本群落组成差异的潜在主成分,二者的主要区别在于 PCA 只基于欧式距离, 而 PCoA 可以基于多种距离矩阵,PCA可以理解为一种特殊的PCoA(基于欧式距离的PCoA就是PCA)。 我们使用R语言ape[23]包, 基于 Bray-Curtis 距离来进行 PCoA 分析, 同时使用R语言vegan[20]包, 基于 Bray-Curtis 距离来进行PERMANOVA分析(即adonis), 以检验分组间微生物群落结构的差异是否显著。

图3-6-14 PCoA 分析结果
- 一个点代表一个样本,点与点空间距离近似物种组成结构的差异程度(Bray-Curtis 距离);
- 两个坐标轴百分比表示两个主成分对样品微生物群落差异(方差)的贡献率。
- 图中PERMANOVA分析的p值用于检验分组间微生物群落结构的差异是否显著
无度量多维标定法(NMDS)统计是一种适用于生态学研究的排序方法,类似于PCoA,通过样本微生物群落结构, 或者基因的组成,可以看出组间差异,组内差异等。NMDS包括一类排序方法, 其设计目的是为了克服以前的排序方法(即线性模型,包括PCoA)的缺点,NMDS的模型是非线性(只考虑距离的秩次,而不在乎距离的大小差异)的, 能更好地反映生态学数据的非线性结构。非度量多维尺度法是一种将多维空间的研究对象 (样本或变量)简化到低维空间进行定位、分析和归类, 同时又保留对象间原始关系的数据分析方法。 适用于无法获得研究对象间精确的相似性或相异性数据,仅能得到他们之间等级(秩次)关系数据的情形。 我们使用R语言vegan[20]包,基于Bray-Curtis 距离来进行 NMDS 分析(metaMDS函数), 同时使用R语言vegan[20]包, 基于 Bray-Curtis 距离来进行PERMANOVA分析(即adonis), 以检验分组间微生物群落结构的差异是否显著。

图3-6-15 NMDS 分析结果
- 一个点代表一个样本,点与点空间距离的排序关系近似物种组成结构的差异程度(Bray-Curtis 距离)的排序关系;
- 图中PERMANOVA分析的p值用于检验分组间微生物群落结构的差异是否显著
- 通常认为stress<0.2时有一定的解释意义;当stress<0.1时,可认为是一个好的排序;当 stress<0.05时,则具有很好的代表性。
3.7 功能定量可视化
3.7.1 基因数目可视化
06.Function/ [功能分析文件夹]
└── [数据库名称]
├── GeneCount [gene数目分析文件夹]
│ ├── bar [gene数目柱形图文件夹]
│ │ └── [数据库功能分类方式]
│ │ └── Count_bar.svg [各功能类别gene数目柱形图]
│ ├── pie [gene数目占比饼图文件夹]
│ │ └── [数据库功能分类方式]
│ │ └── Count_pie.svg [各功能类别gene数目占比饼图]
结果文件夹中有很多数据库功能分类方式的结果,这里只挑一部分说明,其它结果解读类似。
柱形图
柱形图用于展示每个功能类别的gene数量。
CARD Drug Class 是CARD Drug Class数据库的一种功能分类方式

图3-7-1 各CARD Drug Class的gene数量柱形图
若功能类别超过20个,只展示gene数量最高的前20个。
饼图
饼图用于展示每个功能类别的gene数量占比。若类别数目超过20个,只展示gene数量占比最高的前20个

图3-7-2 各CARD Drug Class的gene数量占比饼图
3.7.2 功能丰度可视化
06.Function/ [功能分析文件夹]
└── [数据库名称]
└── Abundance
├── *_abundance_mixed.xls [参考3.4小节解读]
├── *_abundance_stratified.xls [参考3.4小节解读]
├── *_abundance_unstratified.xls [参考3.4小节解读]
├── 01.BasicStatistics [基础统计文件夹]
│ ├── Barplots [百分比堆积柱形图文件夹]
│ │ └── [分组方案名]
│ │ └── [数据库功能分类方式]
│ │ ├── barplot.svg [百分比堆积柱形图]
│ │ └── table.xls [百分比堆积柱形图数据]
│ └── Heatmaps [丰度热图文件夹]
│ └── [分组方案名]
│ └── [数据库功能分类方式]
│ ├── heatmap.svg [丰度热图]
│ └── table.xls [丰度热图数据]
├── 02.GroupComparison [差异比较文件夹]
│ ├── ANOVA_Duncan [ANOVA和Duncan检验结果文件夹]
│ │ └── [分组方案名]
│ │ └── [数据库功能分类方式]
│ │ ├── [分组方案名]_all_feature_anova_results.xls [ANOVA检验结果]
│ │ ├── [分组方案名]_all_significant_feature_duncan_results.xls [Duncan检验结果, ANOVA无显著可缺失]
│ │ ├── [分组方案名]_all_significant_feature_barplot_of_duncan.svg [Duncan检验结果图, ANOVA无显著可缺失]
│ │ └── final_data_for_anova.xls [检验数据(原始丰度log转化,标准化之后)]
│ ├── DunnTest [Kruskal-Wallis和Dunn’s Test检验结果文件夹]
│ │ └── [分组方案名]
│ │ └── [数据库功能分类方式]
│ │ └── DunnTest.xls [Kruskal-Wallis和Dunn’s Test检验结果表格]
│ └── LEfSe [LEfSe分析结果文件夹]
│ └── [分组方案名]
│ └── [数据库功能分类方式]
│ ├── data_for_lefse.xls [LEfSe分析输入文件]
│ ├── lefse.LDA.xls [LEfSe分析结果表格]
│ ├── lefse.normalized.txt [LEfSe标准化后的数据,中间文件]
│ ├── lefse.svg [LEfSe分析结果LDA图]
│ └── SignificantFeatures [差异功能物种来源分层柱形图文件夹]
│ ├── Q5UQG2_stratification_bar.svg [差异功能物种来源分层柱形图]
│ └── table_for_stratification_bar.txt [差异功能物种来源分层柱形图输入表]
└── 03.Correlation [相关性分析结果文件夹]
├── CorrelationHeatmap [相关性热图结果文件夹]
│ └── [数据库功能分类方式]
│ ├── Correlation_heatmap.svg [相关性热图]
│ ├── p_value_matrix.xls [相关系数p值矩阵]
│ └── spearman_rank_correlation_matrix.xls [spearman相关系数矩阵]
└── RDA [CCA/RDA结果文件夹]
└── [分组方案名]
└── [数据库功能分类方式]
├── [分组方案名]_RDA.envfit.xls [RDA各环境因子坐标,解释方差,p值]
├── [分组方案名]_RDA.Factors.PERMANOVA.xls [所有环境因子对样品功能丰度变异的解释方差的p值计算结果]
├── [分组方案名]_RDA_features_location_plot.svg [RDA功能features点图]
├── [分组方案名]_RDA.features.xls [RDA功能features点坐标]
├── [分组方案名]_RDA.Group.PERMANOVA.xls [分组方式对样品功能丰度变异的解释方差的p值计算结果]
├── [分组方案名]_RDA_sample_location_plot.svg [RDA样本点图]
├── [分组方案名]_RDA_sample_location_plot_with_labels.svg [RDA样本点图(带样本名)]
├── [分组方案名]_RDA.sample.xls [RDA样本点坐标]
└── DCA_output.xls [DCA分析各排序轴值]
该部分结果与3.6小节(物种丰度可视化)大体相同, 请参照3.6小节解读功能丰度可视化结果。
这里需要特别说明的是,在LEfSe分析之后,我们挑出存在差异的功能(LDA>2), 用物种来源分层柱形图展示差异功能的物种来源,如图3-7-3

图3-7-3 KEGG数据库差异功能物种来源分层柱形图
四 分析所用软件的版本
| 软件 | 版本 |
|---|---|
| fastp | 0.23.1 |
| BWA | 0.7.17 |
| SortMeRNA | 4.3.6 |
| Trinity | 2.15.1 |
| CDHIT | 4.8.1 |
| QUAST | 5.0.2 |
| diamond | 0.9.14 |
| BASTA | 1.4.1 |
| eggnog-mapper | 2.0.1 |
| RSEM | 1.3.1 |
| DESeq2 | 1.26.0 |
| edgeR | 3.28.1 |
| clusterProfiler | 3.14.3 |
| LEfSe | 1.0.8 |
| dunn.test | 1.3.5 |
| ggpubr | 0.2.4 |
| agricolae | 1.3.3 |
| pheatmap | 1.0.12 |
| vegan | 2.6.0 |
| ggplot2 | 3.3.5.9000 |
| qiime2 | qiime2-2022.2 |
| ape | 5.3 |